Diabetes insipidus
Notes
Introduction
Diabetes insipidus results from a deficiency of, or resistance to, anti-diuretic hormone.
Anti-diuretic hormone (ADH) is a hormone synthesised in the hypothalamus and released by the posterior pituitary. Its release is triggered by a rise in plasma osmolality and its action is to cause reabsorption of water in the kidneys.
The clinical consequence of an inadequate ADH response, as seen in diabetes insipidus, is an inability to concentrate urine with resulting polyuria (increased urine) and polydipsia (increased thirst).
DI is a rare condition with a prevalence estimated to be 1 in 25,000. There are two major forms:
- Central (CDI): caused by reduced secretion of ADH. May be idiopathic (most common) or occur secondary to many conditions including tumours, hypopituitarism or following surgery.
- Nephrogenic (NDI): caused by resistance to the effect of ADH. It can be due to an inherited defect in the gene for the ADH receptor on the X-chromosome. Acquired causes include chronic lithium use and hypercalcaemia.
NOTE: the terms ADH, vasopressin and arginine vasopressin (AVP) are interchangeable.
ADH physiology
ADH release is governed by the plasma osmolality.
Antidiuretic hormone (ADH) is produced by the magnocellular neurons in the paraventricular and supraoptic nuclei of the hypothalamus. It is stored and released by the posterior pituitary in response to rising plasma osmolality. It may also be referred to as arginine vasopressin (AVP) or simply vasopressin.
ADH release begins at a plasma osmolality of around 280 mOsm/kg. Thirst is felt at around 290 mOsm/kg. These processes lead to a reduction in the plasma osmolality.
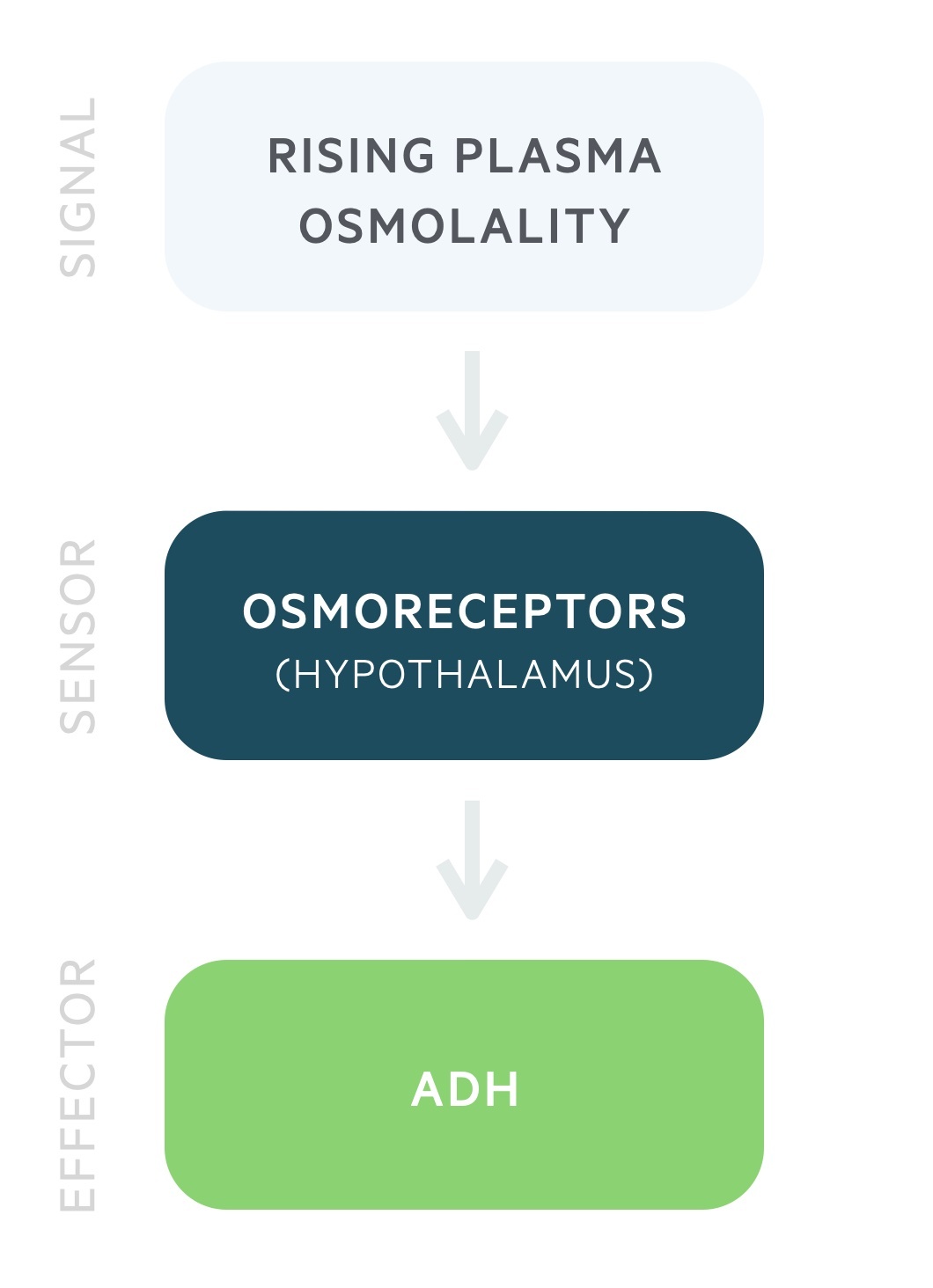
ADH acts on the distal convoluted tubule and collecting duct to increase water reabsorption independent of sodium. It stimulates arginine vasopressin receptor 2 leading to the insertion of aquaporin-2 channels onto the luminal membrane, allowing the free entry of water. ADH also causes vasoconstriction of arterioles.
Aetiology
DI may be divided into two major forms: central and nephrogenic.
Central diabetes insipidus
Central DI is the most common form of DI. It results from the reduced release of ADH. This can occur due to pathology affecting any part of the pathway involved in the synthesis, storage and release of ADH.
Causes of Cental DI include:
- Idiopathic (approx 30-50% of cases, many thought to be due to autoimmune disease)
- Neurosurgery (common, typically transient)
- Trauma
- Familial CDI (e.g.Wolfram syndrome)
- Infection:
- TB
- Abscess
- Meningitis
- Tumours:
- Primary (e.g.craniopharyngioma, hypothalamic tumour)
- Secondary (e.g. lung, leukaemia, lymphoma metastatic spread)
- Infiltrative disease:
- Sarcoidosis
- Langerhan’s cell histiocytosis
- Other (e.g. post-radiotherapy, vascular, hypophysitis)
Nephrogenic diabetes insipidus
Nephrogenic DI occurs in the presence of normal ADH release due to the impaired ability of the kidneys to respond to it. It can be inherited or acquired.
The inherited form occurs due to genetic mutations and presents early in life. Inherited causes include:
- Arginine vasopressin receptor 2 (AVPR2) mutation: is an X-linked recessive condition that accounts for 90% of inherited nephrogenic DI. The AVPR2 gene encodes the arginine vasopressin receptor 2 that is responsible for mediating the anti-diuretic effect of ADH.
- Aquaporin 2 (AQP2) mutation: is normally an autosomal recessive (rarely autosomal dominant) condition that accounts for 10% of inherited nephrogenic DI. The AQP2 gene encodes the aquaporin-2 channels that facilitate the action of ADH.
Acquired nephrogenic DI may occur secondary to a wide range of conditions that impair the ability of ADH to concentrate the urine. Causes include:
- Medications:
- Lithium
- Others (e.g. amphotericin B, demeclocycline, cidofovir)
- Electrolyte disturbance:
- Hypercalcaemia
- Hypokalaemia
- Renal disease:
- Intrinsic renal disease
- Post-obstructive uropathy
Gestational diabetes insipidus
Gestational diabetes insipidus is a rare form of the disease. It is thought to result from the action of the enzyme vasopressinase that is produced by placental trophoblasts. This enzyme breaks down ADH preventing it from carrying out its normal actions.
Clinical features
DI is characterised by polyuria and polydipsia.
Clinical features of DI reflect the inability to concentrate urine. The classic symptoms are:
- Polyuria
- Polydipsia
- Nocturia
The degree of polyuria is highly variable and can reach 10-15L per day in severe cases which can lead to life-threatening dehydration.
Depending on the severity of disease and the patients ability to maintain adequate oral intake signs of dehydration (e.g. dry mucous membranes, prolonged capillary refill, hypotension) may be present.
Clinical features can also reflect the underlying aetiology and as such a thorough history and examination is required.
Investigations
The water deprivation test can be used to diagnose DI.
Bloods
- FBC
- Renal function (check for renal impairment and hypokalaemia)
- Plasma osmolality
- Plasma glucose
- HbA1c
- Bone profile (check calcium)
Urine
- 24-hr urine collection
- Urine specific gravity
- Urine osmolality
Water deprivation test
Protocol
The water deprivation test can be used to diagnose DI. There are many variations on the protocol, here we will give the general outline. The patient fasts and has no fluids from 8.00 am (may also be conducted overnight). Their urine volume, serum and urine osmolality and weight are monitored for up to eight hours.
The patient must be closely monitored and the test stopped if the patient cannot tolerate it or there has been excessive weight loss (in this setting representing dehydration).
Desmopressin (termed DDAVP, a synthetic ADH) may be given at the end of the test (4pm) if the urine osmolality is < 600 mOsm/kg. The patient can then eat and drink with monitoring of urine volume and osmolalities until 8.30pm
Interpretation
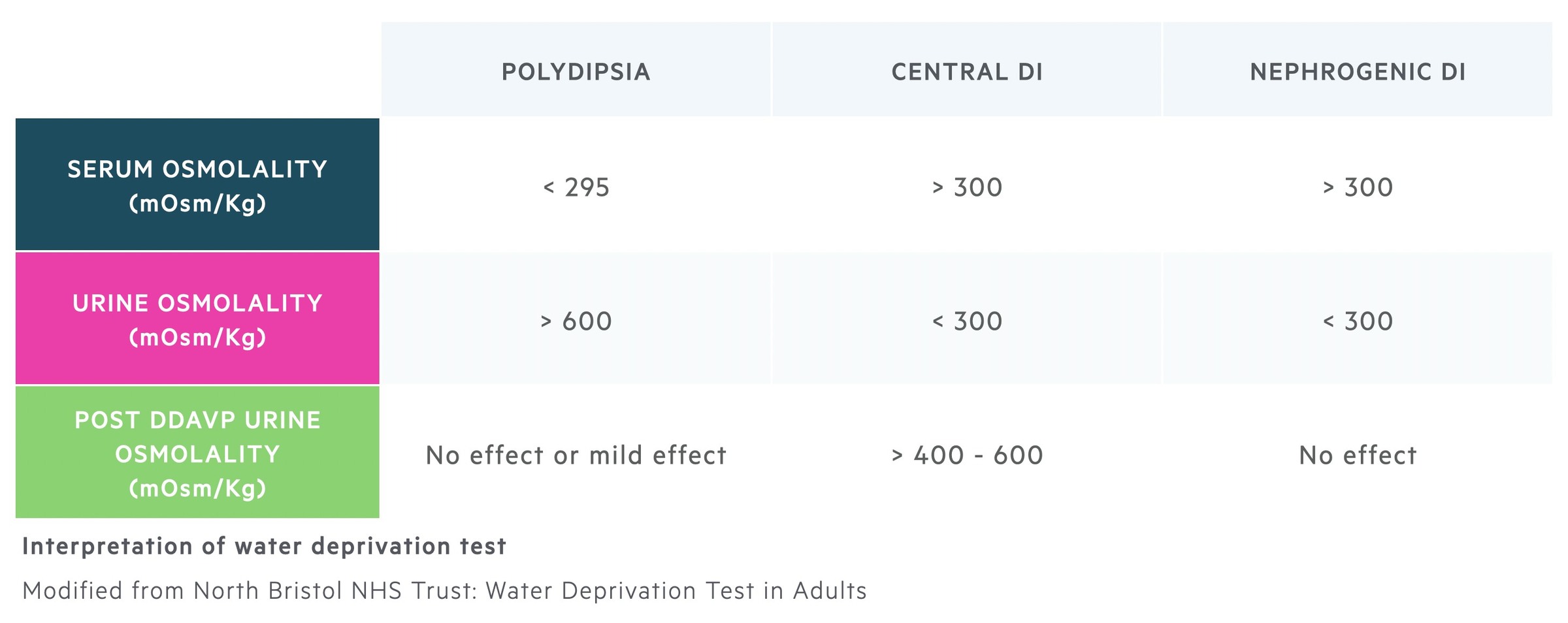
Water deprivation leads to an increase in plasma osmolality. The normal physiological response is to release ADH and concentrate the urine leading to a rise in the urine osmolality. Prior to desmopressin, a urine osmolality > 600mOsm/kg rules out DI and the test can be stopped. A urine osmolality < 400mOsm/kg combined with a raised plasma osmolality is indicative of an inability to concentrate urine.
Urine volumes can also be suggestive of DI. In primary polydipsia (drinking too much water) and normal individuals urine output should decrease during periods of water deprivation. In DI large volumes of dilute urine are passed despite an elevated plasma osmolality.
Following the administration of DDAVP (given to patients with results indicating DI) those with central DI will have a rise in their urine osmolality as the synthetic ADH can concentrate the urine. There will be no effect in patients with nephrogenic DI as the kidneys are not able to respond to the synthetic ADH.
Further investigations
Further testing may be indicated depending on the suspected underlying cause. Examples include MRI pituitary/hypothalamus for tumours or USS renal tract for obstructive uropathy.
Management
Central DI may be treated with the synthetic ADH, desmopressin.
Central DI
Central DI can be treated with the administration of the synthetic ADH, desmopressin. It can be given orally, intranasally or intramuscularly. Compared to endogenous ADH, desmopressin has a longer active duration and does not have vasopressor effects.
One of the risks of desmopressin is water retention resulting in hyponatraemia. This can occur as the synthetic ADH is not suppressible as ADH would be in normal physiology when plasma osmolality falls. As such, particularly at the onset of treatment, close monitoring of plasma osmolality and sodium is needed.
If a treatable underlying cause is identified, this should be appropriately managed.
Nephrogenic DI
If there is a reversible cause (e.g. medication-induced, electrolyte abnormality) then this should be corrected.
Treatment depends on the severity of disease. In milder cases, adequate oral hydration may suffice.
Where medical treatment is required options include:
- Diuretics: thiazide diuretics can be used. It is thought the initial diuresis it causes prompts a hypovolaemia that results in increased sodium and water reabsorption. The effect can be enhanced with amiloride, which can also be effective in lithium-induced DI.
- Desmopressin: in non-hereditary forms of the nephrogenic DI, supraphysiological levels of synthetic ADH may overcome the relative resistance to its actions.
In infants with inherited forms of the disease close specialist care is needed with appropriate hydration and monitoring of electrolytes.
Last updated: April 2022
Have comments about these notes? Leave us feedback