Lynch syndrome
Notes
Overview
Lynch syndrome is an autosomal dominant inherited disorder associated with a high lifetime risk of developing CRC.
Lynch syndrome (LS) is the most common colorectal cancer (CRC) predisposition syndrome accounting for > 3% of CRC cases. This refers to a genetic predisposition to the development of cancer due to certain inherited mutations.
LS is strongly associated with the development of CRC, but also several extra-colonic malignancies. The most common of these is endometrial cancer, but ovarian, gastric, small bowel, urothelial, hepatobiliary and brain malignancies may also be seen.
The estimated lifetime risk of CRC in LS is 40-60%, which depends on mutation type.
Aetiology
Lynch syndrome is an autosomal dominant inherited disorder.
LS is due to a germline mutation (i.e. mutation passed onto our progeny that will be present in every cell in the body) in one of the mismatch repair (MMR) genes:
- MLH1
- MSH2
- MSH6
- PMS2
Genetic variants in MLH1 and MSH2 account > 90% of cases. Rarely genetic mutations in the EPCAM gene can cause LS. Mutations in EPCAM cause silencing of the gene MSH2. The cumulative incidence of CRC differs by the underlying germline mutation, which is greatest for MLH1 and MSH6 mutations.
Mismatch repair pathway (MMR)
The MMR pathway helps in recognition and repair of DNA errors occurring during DNA replication prior to cell division. It helps maintain genomic integrity through correction of base substitutions or small insertion-deletion mismatches.
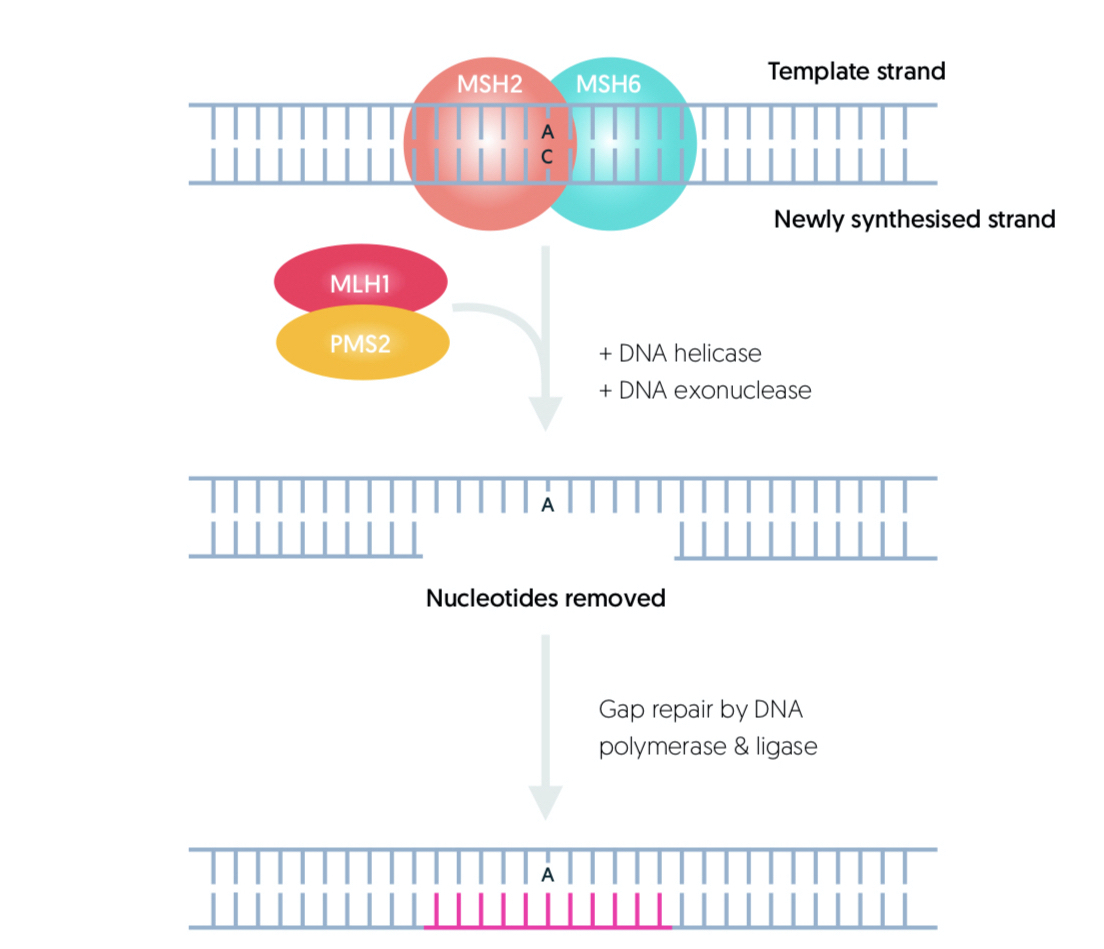
Patients with LS inherit an abnormal MMR gene allele. Subsequent loss of the corresponding allele leads to defective DNA repair promoting tumourigenesis. Patients are at most risk of developing CRC and endometrial cancer.
Pathophysiology
LS predisposes to CRC, which is a heterogenous disease due to a variety of different molecular and genetic alterations.
The development of CRC arises from a variety of molecular pathways, genetic susceptibility and environmental factors that all combine to influence an individuals risk of developing cancer.
Sequential tumour-promoting genetic and epigenetic mutations occur in the colorectal mucosa, which leads to formation of benign adenomas. Overtime, some adenomas progress to invasive carcinoma, which describes the traditional adenoma-to-carcinoma model of colorectal carcinogenesis.
At a molecular level, loss of genomic stability facilitates carcinogenesis through an increasing likelihood of tumour-promoting mutations. In CRC, an average of 80 mutations are found in any single cancer. Microsatellite instability (MSI) due to MMR mutations is one of the pathways for CRC development. It may occur in the presence of a germline mutation such as with LS, or a somatic mutation (i.e. mutation within cells that cannot be passed onto our progeny) that only occurs in the tumour.
Clinical features
Patients with LS may be asymptomatic or may have clinical features due to the development of various cancers.
Constitutional symptoms of malignancy
- Fever
- Anorexia
- Weight loss
- Night sweats
Colorectal cancer
- Abdominal pain
- PR bleeding
- Change in bowel habit
Endometrial cancer
- Post-menopausal bleeding
- Intermenstrual bleeding
- Abnormal uterine bleeding
Other extra-colonic tumours
- Small bowel: nausea and vomiting, GI Bleeding, change in bowel habit
- Pancreatic: back pain, jaundice
- Ovarian: pelvic pain, ascites
- Renal/urinary: flank pain, haematuria
- Gastric: dyspepsia, haematemesis, nausea, vomiting
Muir-Torre syndrome
This refers to a variant of LS, which is characterised by sebaceous tumours and cutaneous keratoacanthomas in addition to LS-associated tumours. A Keratoacanthoma is a rapidly-growing skin tumour that looks like a squamous cell carcinoma.
Turcot syndrome
This is a historical term, which refers to the presence of hereditary CRC and brain tumours, particularly medulloblastomas and gliomas. It is now known that both LS and familial adenomatous polyposis (FAP), another CRC predisposition syndrome, are associated with the development of gliomas and medulloblastomas
Amsterdam criteria
The Amsterdam criteria is used to define LS based on family history.
Traditionally, the Amsterdam criteria (AC) was used to determine patients with a family history that is suggestive of LS, which requires further diagnostic testing.
AC can be remembered using the 3-2-1 rule:
- ≥3 family members affected (colorectal cancer or endometrial cancer)
- ≥2 two generations (e.g. parents and grandparents or parents and children)
- ≥1 family member diagnosed at young age (before 50 years old)
The AC has a low sensitivity for the diagnosis of LS. This means a significant proportion of patients with LS will not meet the criteria. Consequently, further assessment of tumour biology is needed to establish whether genetic testing is warranted. This means looking at LS-associated tumours for microsatellite instability, which is the hallmark of LS.
Testing for microsatellite instability
The diagnostic work-up for LS starts with MSI testing of a colorectal tumour using PCR or immunohistochemistry.
Tumours with MMR deficiency are characterised by MSI. Microsatellites are repetitive pieces of DNA prone to errors during replication. In MMR defects, microsatellites accumulate errors leading to changes in the size of their repetitive sequences.
Two methods for determining MSI are PCR-based and immunohistochemistry (IHC).
Immunohistochemistry
IHC is a common method that utilises monoclonal antibodies to identify MMR proteins in the tumour. An MMR gene mutation leads to loss of expression of the corresponding protein. The presence of MMR protein loss warrants genetic testing for a germline mutation that would imply LS.
In some cases, MMR protein loss on IHC warrants further somatic tumour testing (this means testing for common mutations associated with sporadic colorectal cancers). This is because some sporadic tumours show MSI without an underlying germline mutation (i.e. an MMR mutation has occurred in the tumour only). These are associated with common somatic mutations such as BRAF and need to be distinguished from LS. This may be considered in an elderly patient with MSI-associated CRC and no significant family history.
PCR-based testing
Polymerase chain reaction (PCR) can be used to test for MSI. This technique essentially involves the amplification of DNA to look at defects in microsatellites and compare between tumour DNA and normal colorectal mucosal DNA. If there is a marked different the tumour is reported to have high level of MSI (MSI-H). The presence of MSI-H warrants further genetic testing for LS. IHC is generally preferred to PCR-based testing.
Bethesda guidelines
Originally, decisions for MSI-testing were based on the Bethesda guidelines, which have a number of criteria focusing on CRC, age and family history. If a single point was met, the tumour underwent MSI-testing. However, in line with NICE guidelines, all CRC should undergo empirical MSI-testing.
Diagnosis
A formal diagnosis of LS is made on genetic testing for a pathological mutation in one of the MMR genes.
If LS is suspected based on MSI testing of a CRC, or based on family history, then formal genetic testing is warranted to look for a pathological mutation in one of the MMR genes.
Patients may be referred for genetic testing by several routes:
- Characteristic family history of LS (i.e. meets Amsterdam Criteria)
- Confirmed family history of LS
- Personal or family history of CRC with MSI
In patients with a family history of CRC or endometrial cancer with MSI, then the affected relative should be referred for genetic testing. If this is not possible (i.e. estranged relatives or passed away) then genetic testing may be offered to the current patient if the clinical suspicion is high. Local genetic guidance is usually followed.
The risk, benefits and limitation of genetic testing should always be discussed with the patient
Surveillance
Patients with confirmed LS, or in those where it is highly suspected but testing is unavailable, require colonoscopic surveillance.
Colonoscopic surveillance
Colonoscopic surveillance refers to regular colonoscopies to assess the colorectal mucosa. The aim of surveillance is to prevent CRC or detect it at a much earlier stage. Colonoscopic surveillance in LS has shown to reduce the risk of developing and dying from CRC, which is based on several observational cohort studies.
The British Society of Gastroenterologists (BSG) have produced guidelines for the management of hereditary colorectal cancer, which includes advice on colonoscopic surveillance in patients with a family history of CRC and those with known hereditary syndromes.
In LS, patients require colonoscopic surveillance at two yearly intervals:
- MLH1/MSH2 mutations: 2 yearly colonoscopy from 25 years old
- MSH6/PMS2 mutations: 2 yearly colonoscopy from 35 years old
Extra-colonic surveillance
Patients with LS are at risk of extracolonic malignancies. At present, insufficient data exists to support surveillance for these cancers outside a clinical trial.
Patients should be screened for Helicobacter pylori and undergo eradication therapy as appropriate. This is because of the high proportion of ‘intestinal type’ gastric cancer in LS. 'Intestinal type' is strongly linked with H. pylori infection.
Management
The management of LS involves the early recognition and treatment of premalignant and malignant lesions.
Colorectal cancer
High-risk polyps (small benign mucosal growths) and CRC detected on surveillance, or ad hoc in patients presenting with new symptoms, should be discussed at a multi-disciplinary team meeting and managed accordingly. Management strategy is similar in patients with LS and sporadic CRC. However, the risk of metachronous cancer (occurring at a different time) needs to be taken into consideration in LS and patients will usually require ongoing surveillance following surgery.
Extracolonic tumours
The management of extracolonic tumours should follow the same pathways for sporadic tumours and be managed accordingly.
Chemoprophylaxis
Aspirin is used as a chemoprophylactic agent in patients with LS, which has been shown to significantly reduce the risk of CRC in the CAPP2 randomised controlled trial. The optimum dose needs to be established, which is being assessed in ongoing trials. At present, patients should be offered low-dose aspirin to reduce CRC risk.
Genetic services
Patients require referral to specialist family cancer services with access to a genetic counsellor and a clinician with experience in hereditary CRC. These services offer genetic testing, family screening and provide appropriate specialist knowledge for LS with awareness of patient/support organisations and ongoing research opportunities.
Last updated: March 2021
Have comments about these notes? Leave us feedback