Beta thalassaemia
Notes
Overview
Thalassaemia refers to a group of disorders characterised by reduced or absent globin chain production.
Thalassaemia is one of the haemoglobinopathies, which refers to a group of autosomal recessive inherited disorders that affect the globin chains that form the protein component of haemoglobin.
Haemoglobinopathies
Haemglobinopathies can be broadly divided into two types:
- Haemoglobin variants: mutant forms of haemoglobin that affect the structure. Sickle cell disease is most well recognised.
- Thalassaemia: reduced or absent globin chain production due to underlying mutations.
Thalassaemia
Thalassaemia can be further divided depending on the type of globin chain affected:
- Alpha thalassaemia: reduced or absent production of the alpha globin chains
- Beta thalassaemia: reduced or absent production of the beta globin chains
Haemoglobin
Haemoglobin is the main oxygen-carrying molecules within our red blood cells.
Haemoglobin (Hb) is essential for the transport of oxygen around the body. It is composed of four globin chains and four heme molecules, which are the actual oxygen-binding structures that contain iron.
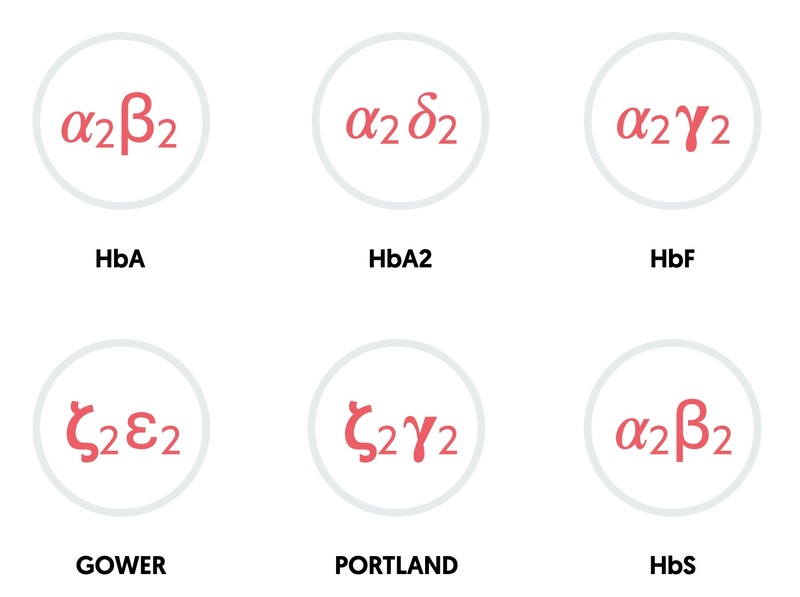
Type of haemoglobin
The combination of different globin chains determines the type of haemoglobin.
- HbA: two alpha, two beta (95-98% in adults)
- HbA2: two alpha, two delta (2-4% in adults)
- HbF: two alpha, two gamma (fetal haemoglobin: 0.8-2% in adults)
- Gower: two zeta, two epsilon (embryonic haemoglobin)
- Portland: two zeta, two gamma (embryonic haemoglobin)
Genetics
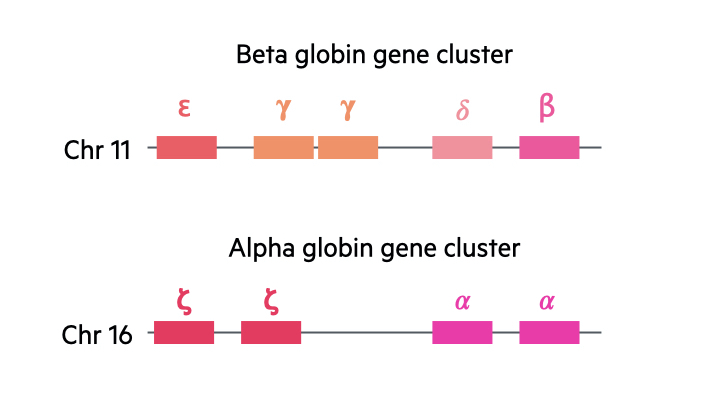
In the human body, there are two major gene clusters important for the synthesis of globin chains in the formation of haemoglobin:
- Alpha globin gene cluster: located on chromosome 16. Contains the embryonic globin genes zeta and two copies of the alpha globin gene (alpha-1 and alpha-2).
- Beta globin gene cluster: located on chromosome 11. Contains the embryonic globin gene epsilon, fetal globin genes and the adult beta globin and delta globin gene.
We contain four alpha globin genes and two beta globin genes. With the alpha genes, two are inherited paternally and two inherited maternally. With the beta globin genes, one is inherited from each parent.
Mutations types
When we look at abnormal mutations seen in thalassaemia, we use the following denotation:
- Mutation leading to absent production (0)
- Mutation leading to reduced production (+)
Epidemiology
Beta thalassaemia is most common among Mediterranean, African, and Southeast Asian populations.
In the UK, it is estimated that around 1000 people have beta thalassaemia major (the most severe form) with around 300,000 carriers. The condition is most commonly seen in populations around the Mediterranean, Africa and Southeast Asia.
Beta thalassaemia in patients of Mediterranean origin are most likely to present with anaemia. This is because the mutation most commonly seen in this population leads to absent production of the beta globin chain.
Aetiology
In beta thalassaemia, mutations in the beta globin genes lead to reduced or absent production.
We each contain two copies of the beta globin gene. One is inherited paternally and one maternally. Beta thalassaemia is considered an autosomal recessive condition, but the clinical phenotype (i.e. anaemia) can be seen with even one abnormal beta globin gene. However, the severe form of the condition requires two abnormal genes.
Genetic mutations
More than 200 mutations have been identified in the beta globin gene that can cause a reduced or absent production of the beta globin chain.
Mutations seen can include:
- Deletions
- Genetic rearrangements
- Mutations affecting transcription (due to mutations that affect promotors or enhancers)
- Mutations affecting splicing (splicing is form of mRNA processing that removes introns)
- Premature stop codons (affects mRNA translation leading to truncated proteins)
- Other rarer mechanisms
Types of beta thalassaemia
There are three types of beta thalassaemia known as minor, intermedia and major.
- Beta thalassaemia minor (b+/b OR b0/b): this is also known as beta thalassaemia trait. Patients have one abnormal beta globin gene, which may be associated with reduced production (+) or absent production (0). Usually asymptomatic with mild anaemia.
- Beta thalassaemia intermedia (b+/b+): this refers to patients with two abnormal beta globin genes. Both lead to a reduced production of beta globin chains. These patients are usually non-transfusion dependent.
- Beta thalassaemia major (b+/b0 OR b0/b0): this refers to patients with minimal or absent beta globin chain production due to mutations in both genes. These patients are transfusion dependent.
Pathophysiology
The absence of beta globin chain production leads to an alpha/beta globin chain imbalance and chronic haemolytic anaemia.
Beta thalassaemia minor or trait is typically asymptomatic with mild microcytic anaemia. Patients with beta thalassaemia major have severe disease with chronic haemolytic anaemia and ineffective erythopoiesis.
Beta thalassaemia major typically presents after 6 months of age when there is a change from fetal haemoglobin (HbF) to normal adult haemoglobin (HbA). The reduced beta globin chain production leads to an imbalance in alpha and beta chains that causes ineffective erythropoiesis (i.e. formation of new red blood cells) with haemolysis in the bone marrow and peripheral blood. This is coupled with reduced red blood cell survival. In addition, there is increased iron absorption leading to overload, which is compounded by regular transfusions.
One of the hallmarks of beta thalassaemia is extramedullary haematopoiesis, which refers to haematopoiesis occurring outside the medulla of long bones. Typical sites include the spleen, liver and unusual bones including the skull. Despite the increase in haematopoiesis sites, it is still ineffective due to the absence of beta globin chains. There is an increased production of HbA2 that contains the delta chain and HbF that contains the beta-like gamma chain. However, this increased production is not sufficient to meet the demands of the body. In addition, excess alpha chains leads to the formation of unstable alpha tetramers that precipitate in red blood cells causing premature destruction.
Collectively, this results in chronic, transfusion-dependent anaemia.
Clinical manifestations
Beta thalassaemia causes a range of clinical manifestation due to haemolysis, extramedullary haematopoiesis and iron overload.
Anaemia
The severity of anaemia and subsequent symptoms depends on the beta thalassaemia type:
- Minor/trait: usually asymptomatic with mild microcytic anaemia with haemoglobin counts > 100 g/L.
- Major: severe transfusion-dependent anaemia. Features of pallor, dyspnoea, dizziness, lethargy. Untreated, haemoglobin may be as low as 30-40 g/L.
Jaundice
Due to chronic haemolytic anaemia of abnormal red blood cells, patients may have evidence of a unconjugated hyperbilirubinaemia and gallstones from pigment stones.
Skeletal changes
Boney changes are common in patients with beta thalassaemia major due to ineffective erythropoiesis and extramedullary haematopoiesis.
- Facial deformity: frontal bossing, maxilla overgrowth, prominence of upper incisors, ‘chipmunk’ facies, dental malocculsion.
- Body habitus changes: typically short limbs due to early fusion of epiphyses. Skull, pelvis, ribs and spinal changes may be seen
- Osteopaenia/osteoporosis: osteopaenia may be seen in 45% and osteoporosis in 15-50% of patients, which can lead to pathological fractures. Linked to bone marrow space widening. This can lead to a ‘hair-on-end’ appearance in the skull, seen on x-ray.
- Boney pain
Iron overload
Ineffective erythropoiesis leads to an increase in iron absorption from the gastrointestinal tract that is compounded by regular blood transfusions. Iron chelation therapy is needed from an early age to prevent complications. Disorders associated with iron overload include:
- Growth impairment
- Hepatic impairment
- Cardiac failure
- Cardiac arrhythmias
- Endocrine disorders: hypogonadism and hypothyroidism (pituitary iron deposition) and diabetes mellitus (pancreatic iron deposition).
Other abnormalities
- Pulmonary: at risk of obstructive and restrictive defects. Some patients may develop pulmonary hypertension
- Thrombotic: beta thalassaemia intermedia and major are associated with a hypercoagulable state.
- Leg ulcers
Diagnosis & investigations
Anaemia in beta thalassaemia usually develops from 6-12 months of age.
Most patients with beta thalassaemia minor will be completely asymptomatic and not realise they have the underlying condition. Beta thalassaemia intermedia/major typically presents from 6 months of age with the usual switch from HbF to HbA.
The diagnosis of beta thalassaemia is usually obvious with the presentation of severe anaemia in the first year of life. Initial testing can confirm the presence of chronic haemolytic anaemia and diagnostic testing is important to confirm beta thalassaemia.
Initial testing
- Full blood count: looking for low haemoglobin and low mean corpuscular volume (MCV). RBC count is typically increased with a low reticulocyte count.
- Blood film: typically shows hypochromic, microcytic red blood cells. Abnormal red cell morphology is may be seen. May see red blood cell inclusions due to precipitated globin chains.
- Liver function tests: unconjugated hyperbilirubinaemia
- Haemolysis screen: Lactate dehydrogenase (raised), haptoglobin (reduced), direct antiglobulin test (Negative, non-immune haemolysis)
- Iron studies: these are important to exclude iron-deficiency (cause of microcytic anaemia) and to determine whether iron overload is present.
Diagnostic testing
Diagnostic testing can be used to confirm the presence of beta thalassaemia. It involves haemoglobin analysis and genetic testing.
- Haemoglobin analysis: completed using haemoglobin electrophoresis or high-performance liquid chromatography (HPLC). Electrophoresis causes different types of haemoglobin to separate into bands. HPLC is an alternative method of determining the types of haemoglobin in blood.
- Genetic testing: DNA testing provides a definite and precise diagnosis of beta thalassaemia. It is able to determine the type of mutation present.
On haemoglobin analysis, patients with beta thalassaemia will have an increased proportion of HbA2 and HbF due to the absence of beta globin chains. Even in beta thalassaemia minor, there will be an elevation in HbA2.
Antenatal screening
Screening for thalassaemia is offered to all pregnant women within the UK.
Antenatal screening is offered to all pregnant women within the UK. It involves concurrent assessment of different haemoglobinopathies (i.e. thalassaemia and haemoglobin variants) at 10 weeks gestation. Family origin questionnaires are essential as part of the screening programme to determine the risk of being a carrying of a haemoglobinopathy gene.
In beta thalassaemia, the level of HbA2 is quantified. Levels of HbA2 >3.5% is suggestive of being a beta thalassaemia carrier and further analysis of the father is required to determine the risk of beta thalassaemia in the fetus.
Screening for alpha thalassaemia is more difficult. This is because detection of alpha thalassaemia minima (aa/a-) or alpha thalassaemia trait (a-/a-) can only be completed by DNA testing as there are no specific biomarkers. Therefore, in pregnant mothers if the mean corpuscular haemoglobin is < 25 pg and they are from a high risk area (e.g. China, Southeast asian, etc) then testing of the biological father should be offered. If the biological father is also suspected of having alpha thalassaemia then both the mother and biological father should undergo DNA analysis.
Public Health England (PHE) have produced clear guidelines on the sickle cell and thalassaemia screening programme.
Management
Beta thalassaemia requires lifelong adherence to transfusion programmes and iron chelation therapy.
The mainstay of treatment in beta thalassaemia major is chronic red blood cell transfusions to treat severe, asymptomatic anaemia. In addition, patients require monitoring for complications associated with iron overload and iron chelation therapy. Rarely, newer disease modifying agents and haematopoietic stem cell transplantation, which is potentially curative, can be considered. However, this is beyond the scope of these notes.
Chronic blood transfusions
The aim of chronic blood transfusions is to keep the haemoglobin level at a constant stable state and reduce complications associated with ineffective and extrameullary haematopoiesis.
Patients will have pre-transfusion and post-transfusion haemoglobin targets. This usually involves regular transfusions every 2-3 weeks. Extended cross-matching is usually required to prevent alloimmunisation, which refers to development of an immune response to red blood cell antigens.
Iron overload and chelation therapy
Iron overload is inevitable in patients with beta thalassaemia major and it can cause significant morbidity due to organ toxicity. Therefore, iron levels need to be monitored regularly using ferritin +/- formal iron studies and magnetic resonance imaging can be used to quantify the amount of iron deposition in organs. This is particularly important for hepatic and cardiac tissue.
Patients will usually be started on iron chelation therapy in childhood. There are various indications about when to start chelation therapy. Typical agents include Deferasirox, Deferoxamine and Deferiprone. These chelators bind iron and increase excretion through urine and/or faeces.
Additional management
Splenectomy can be considered in selected patients with symptomatic splenomegaly, hypersplenism (decrease in blood counts) or a dramatic increase in transfusion requirements. Other treatments and monitoring depend on the complications associated with thalassaemia (e.g. hepatic disease, cardiac disease).
Prognosis
Patients with beta thalassaemia major have a reduced life expectancy.
Patients with beta thalassaemia major will not survive without lifelong transfusion programmes. Major mortality is associated with chronic anaemia and iron overload. Life-expectancy depends on the extent of complications associated with iron overload and concordance with life-long treatment. For example, significant cardiac involvement leads to earlier mortality.
Beta thalassaemia minor has no impact on patient morbidity or mortality.
Last updated: April 2021
Have comments about these notes? Leave us feedback