Sickle cell disease
Notes
Introduction
Sickle cell disease refers to a group of conditions that are characterised by inheritance of sickle haemoglobin.
Sickle cell disease (SCD) is one of the most common inherited disorders. It is caused by inheritance of an abnormal beta globin gene, which leads to sickle haemoglobin. It is classified as one of the haemoglobinopathies.
Haemoglobinopathies
Haemoglobinopathies refer to a group of genetic diseases that affect the structure of haemoglobin. They can be broadly divided into two types:
- Haemoglobin variants: mutant forms of haemoglobin that affect its structure. Sickle cell disease is the most recognised.
- Thalassaemia: reduced or absent globin chain production due to underlying mutations.
Haemoglobin variants
There are numerous haemoglobin variants across the world (>1000) that affect red blood cells in different ways.
- Globin chains: some variants affect the alpha chain, whereas others affect the beta chain.
- Clinical severity: some variants are clinically silent, whereas others cause clinical manifestations (e.g. haemolytic anaemia).
- Frequency: sickle haemoglobin is the most common variant. Others are extremely rare and only seen in small populations.
Sickle cell disease
Sickle cell haemoglobin is a haemoglobinopathy inherited in an autosomal recessive pattern and has the highest prevalence in West Africa. The condition usually presents in childhood and is characterised by abnormal, rigid, sickle shaped red blood cells (erythrocytes) leading to the name ‘sickle cell anaemia’.
It is a multi-system disorder due to widespread organ dysfunction from red blood cell ‘sickling’ leading to chronic vaso-occlusion (i.e. blockage of vessels).

Terminology
Sickle cell anaemia refers to the inheritance of two abnormal sickle haemoglobin (HbSS).
The terminology associated with sickle haemoglobin is important to highlight. It depends on the type of haemoglobin variant inherited, the number of variants inherited (i.e. homozygous or heterozygous) and whether there is co-inheritance with a thalassaemia gene.
- Sickle cell anaemia (e.g. HbSS): inheritance of two abnormal sickle genes (one paternally and one maternally).
- Sickle cell disease (e.g. HbSC): inheritance of one abnormal sickle gene and a second haemoglobin variant of the beta chain that causes sickling. For example, haemoglobin C.
- Sickle cell trait (e.g. HbS): inheritance of one abnormal sickle gene (either paternally or maternally). Also known as a sickle carrier.
- Sickle-thalassaemia (e.g. HbSβ0): inheritance of one abnormal sickle gene and one abnormal thalassaemia gene (e.g. alpha or beta). Severity depends on the thalassaemia gene inherited.
Haemoglobin
Haemoglobin is the main oxygen-carrying molecule within our red blood cells.
Haemoglobin (Hb) is essential for the transport of oxygen around the body. It is composed of four globin chains and four heme molecules, which are the actual oxygen-binding structures that contain iron.
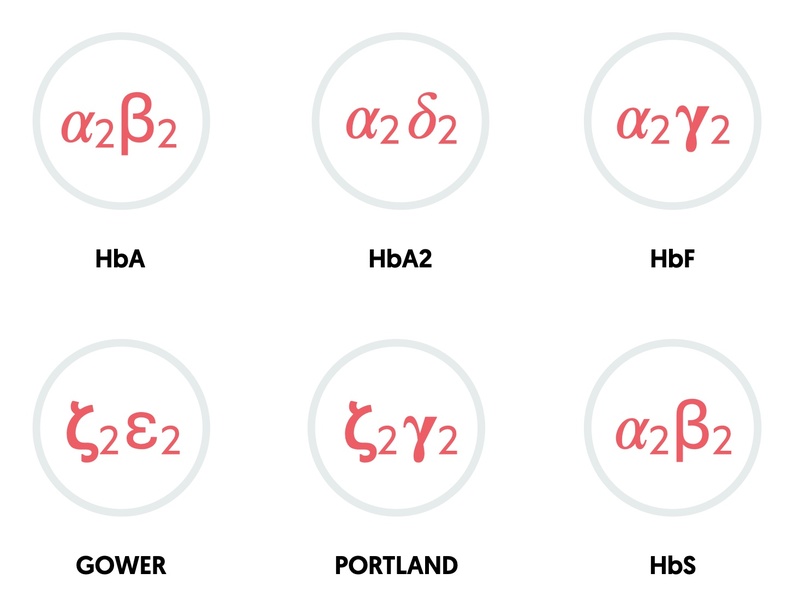
Type of haemoglobin
The combination of different globin chains determines the type of haemoglobin.
- HbA: two alpha, two beta (95-98% in adults)
- HbA2: two alpha, two delta (2-4% in adults)
- HbF: two alpha, two gamma (foetal haemoglobin: 0.8-2% in adults)
- HbS: two alpha, two sickle (abnormal beta)
- Gower: two zeta, two epsilon (embryonic haemoglobin)
- Portland: two zeta, two gamma (embryonic haemoglobin)
NOTE: patients with sickle cell trait will have various amounts of both HbS (~40%) and HbA (~60%).
Genetics
In the human body, there are two major gene clusters important for the synthesis of globin chains.
- Alpha globin gene cluster: located on chromosome 16. Contains the embryonic globin genes zeta and two copies of the alpha globin gene (alpha-1 and alpha-2).
- Beta globin gene cluster: located on chromosome 11. Contains the embryonic globin gene epsilon, fetal globin genes and the adult beta and delta globin genes.
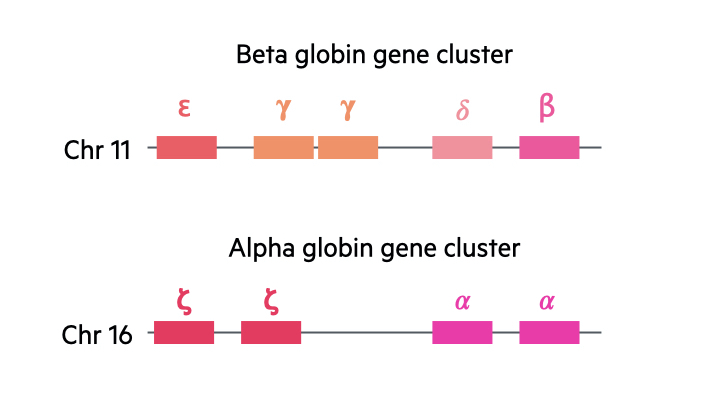
We contain four alpha globin genes and two beta globin genes. With the alpha genes, two are inherited paternally and two inherited maternally. With the beta globin genes, one is inherited from each parent.
Nomenclature
When we look at abnormal mutations seen in thalassaemia, we use the following denotation:
- Mutation leading to absent production (0)
- Mutation leading to reduced production (+)
With haemoglobin variants, there are numerous different mutations that are associated with specific names:
- Haemoglobin S
- Haemoglobin C
- Haemoglobin E
- Haemoglobin Philly
- Haemoglobin D-Punjab
Epidemiology
In the UK, SCD affects approximately 1 in 2000 live births.
SCD is one of the most common inherited diseases worldwide, which predominantly affects patients of African ancestry. The disease is most frequency found in sub-Saharan Africa, with a gene frequency up to 30% in some regions.
The high prevalence of SCD in Africa is speculated to be due to a survival advantage for heterozygous carriers (i.e. one normal gene and one sickle gene) in malarial regions. Other commonly affected regions include Eastern Mediterranean, Middle East, India, Caribbean, South and Central America.
In the UK, there is estimated to be 12500 patients with SCD (as per British Society of Haematologists - 2018). There is a 1:1 sex ratio and symptoms usually begin in the second half of the first year of life when the levels of foetal haemoglobin normally begin to fall.
Aetiology
Sickle haemoglobin is due to a point mutation in the beta-globin gene.
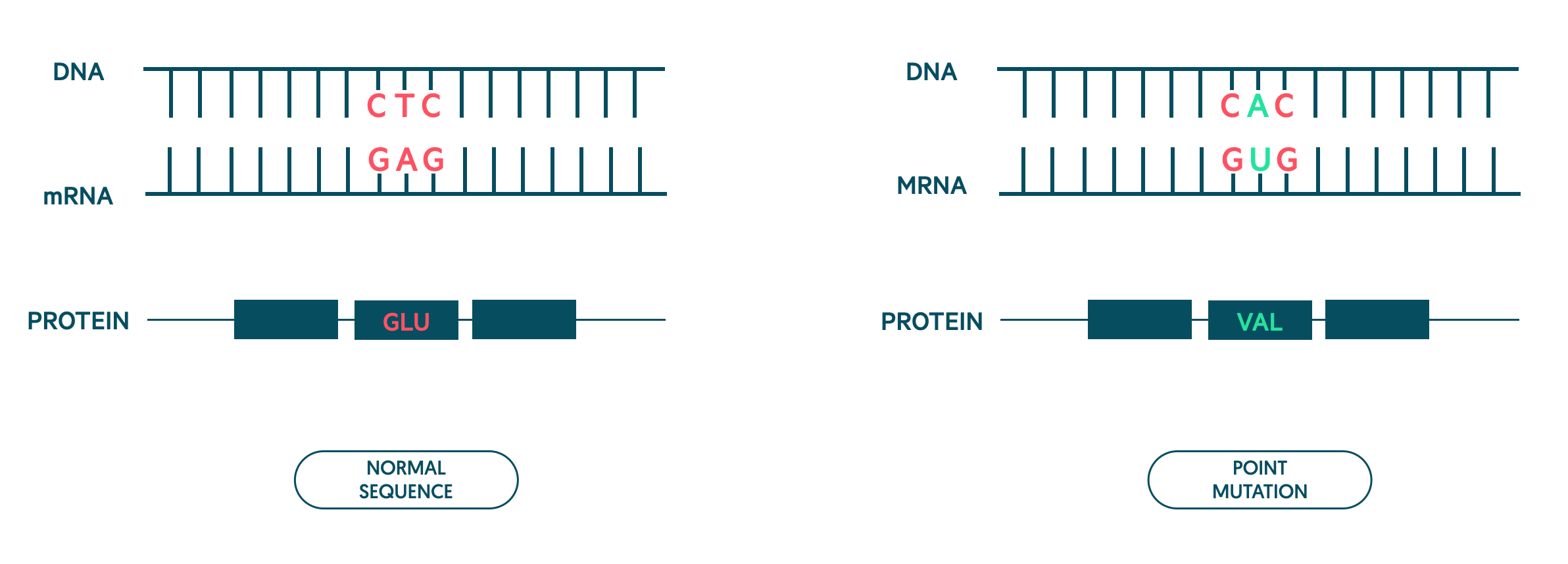
Genetic mutation
Sickle haemoglobin is due to a point mutation in the beta-globin gene located on chromosome 11. A point mutation refers to a single base substitution. In sickle haemoglobin, this affects the sixth amino acid that causes the amino acid glutamic acid to be converted to valine.
Inheritance
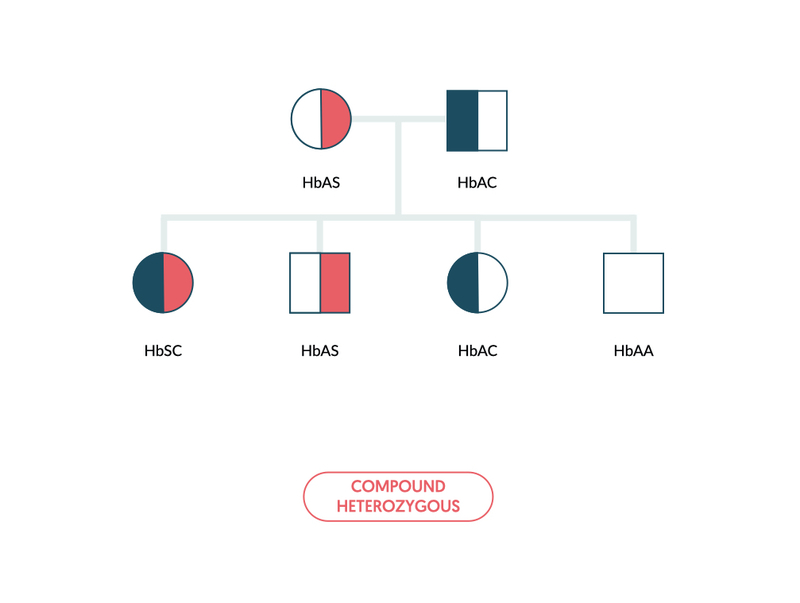
Sickle haemoglobin is inherited in an autosomal recessive pattern. Inheritance of two abnormal sickle haemoglobin genes is known as sickle cell anaemia and patients are homozygous (e.g. HbSS).
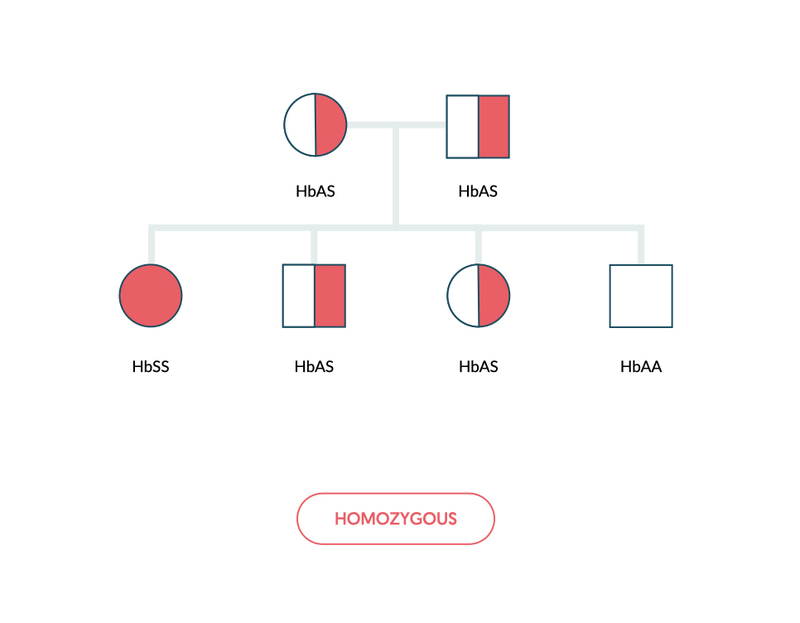
Inheritance of sickle haemoglobin alongside another haemoglobin variant is referred to as compound heterozygous (e.g. HbSC). This refers to the presence of two or more different recessive alleles.
Pathophysiology
The abnormal sickle haemoglobin polymerises at low oxygen tension, which distorts its shape and leads to vessel occlusion.
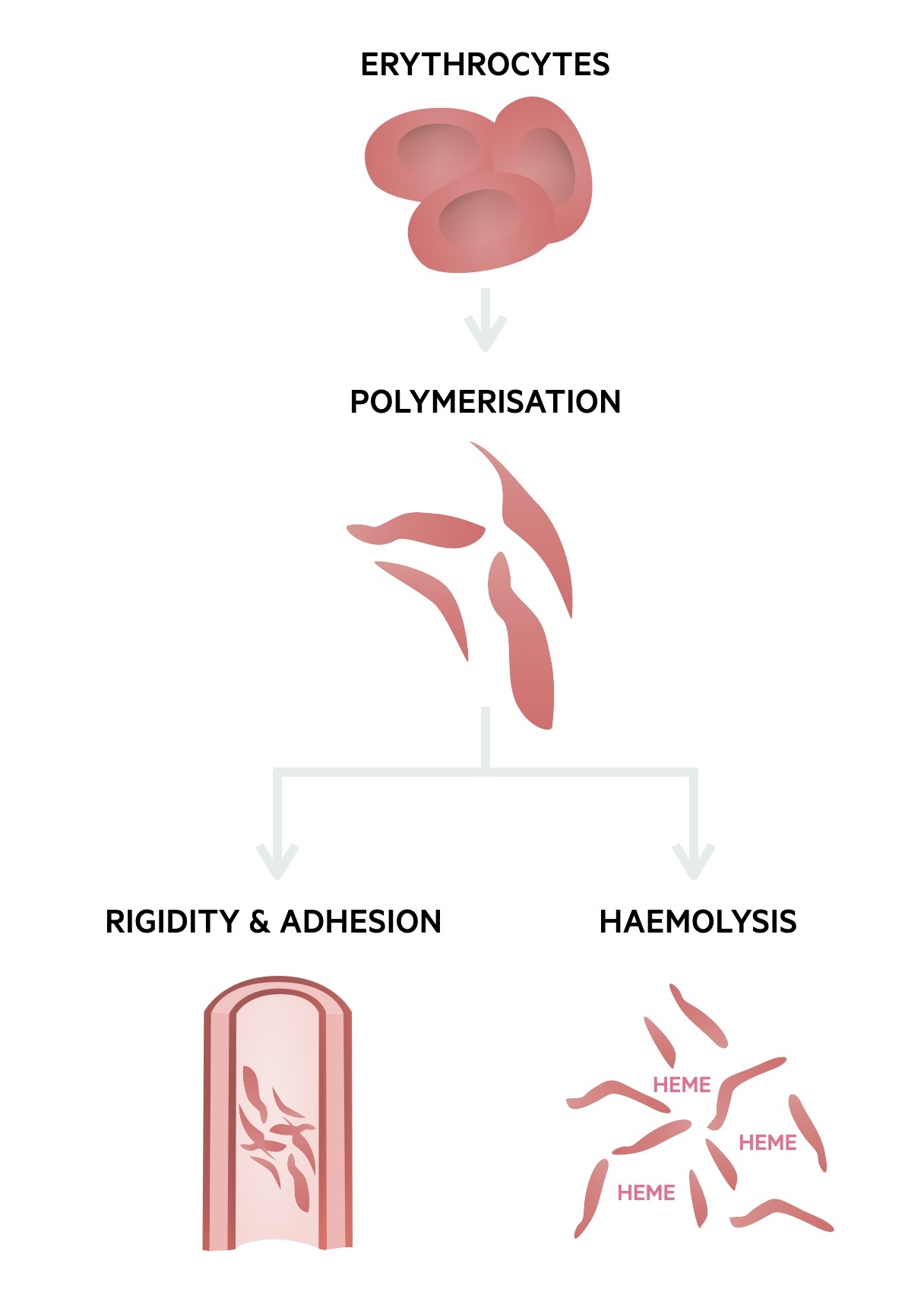
The hallmark of SCD is the formation of sickle-shaped red blood cells that occur due to polymerisation of sickle haemoglobin when placed under low oxygen tension. This damages red blood cells leading to chronic haemolysis and clustering that results in occlusion of blood vessels (known as vaso-occlusion). Haemolysis shortens the lifespan of red blood cells to ~10-20 days compared to 120 days in normal red cells.
Sickling process
Sickle haemoglobin can carry oxygen, but once offloaded to tissue, it begins to precipitate forming semi-solid aggregates. This greatly alters the red blood cell shape, which makes them rigid and at risk of damage. Repeated deoxygenation cycles cause permanent damage and haemolysis (destruction of red blood cells within 100 days).
This sickling process is greatly increased by low oxygen levels, dehydration, concurrent illness (e.g. infections), cold exposure and acidosis.
Vaso-occlusion
Sickling and occlusion of blood vessels is known as vaso-occlusion. It causes tissue ischaemia (inadequate blood supply) that leads to infarction (tissue death from ischaemia). The actual molecular and cellular pathogenesis of vaso-occlusion is more complex than just vessel occlusion. It involves endothelial dysfunction, altered nitric oxide levels (dilates vessels), hypercoagulability and neutrophil activation.
One of the hallmarks of vaso-occlusion is an acute painful episode (also known as ‘painful crisis’). The consequences of vaso-occlusion include:
- Acute painful episodes (i.e. painful crisis)
- Acute chest syndrome (i.e. chest crisis)
- Renal infarction
- Bone infarction or dactylitis (inflammation of a digit)
- Myocardial infarction
- Stroke
- Venous thromboembolism
- Priapism (persistent, painful erection)
Other disease mechanisms
Due to their abnormal shape, red blood cells can get sequestrated (i.e. trapped) within the liver and spleen leading to marked anaemia. If this occurs acutely, if can lead to life-threatening anaemia.
Patients are at increased risk of infection, which is multi-factorial. There is evidence of hyposplenism and/or asplenism due to recurrent infarction, which increases the risk of bacterial infections (particularly encapsulated bacteria). In addition, patients are at increased risk of viral infections (e.g. Parvovirus B19), which can occasionally cause aplastic crises (i.e. transient arrest of erythropoiesis).
Clinical presentation
Sickle cell disease is a multi-system disorder that leads to a variety of acute and chronic complications.
The clinical consequences of SCD are broadly divided into acute and chronic complications. Acute complications include infections, severe anaemia and vaso-occlusive phenomena. Chronic complications occur due to long-standing organ-damage and may be exacerbated by treatments.
Infections
Patients are at increased risk of bacterial infections, particularly encapsulated bacteria such as Pneumococcal, Meningococcal and Haemophilus. This is due to development of hypo-/asplenism. Clinical features associated with infections are usually organ specific and may include fever, headache, cough, dysuria, abdominal pain and/or rash.
Patients are also at risk of viral infections such as parvovirus and influenza. Features of coryzal symptoms with a blocked/runny nose, myalgia, arthralgia and/or fever may be present.
Anaemia
Patients usually have chronic, compensated anaemia due to haemolysis. However, they can develop severe symptoms due to acute falls in haemoglobin (Hb). Three major conditions lead to an acute drop:
- Splenic sequestration: life-threatening condition, usually in children. Acute fall on Hb due to pooling of red blood cells in the spleen. Presents with rapidly enlarging spleen, features of anaemia +/- hypovolaemic shock
- Hyperhaemolysis: sudden exacerbation of haemolysis. May be associated with acute vaso-occlusive crises, due to excess transfusions and development of alloimmunisation (immune response to foreign antigens) to different red blood cell antigens or coexistence of glucose-6-phosphate dehydrogenase (G6PD) deficiency.
- Aplastic crisis: transient arrest of erythropoiesis, usually induced by infection (e.g. parvovirus B19). Evidence of erythropoiesis recovery is usually seen after 2-14 days through increase in reticulocyte formation.
Vaso-occlusive phenomena
As discussed, numerous organs can be affected by vaso-occlusive phenomena. Two of the most clinically relevant vaso-occlusive phenomena are acute painful episodes, also known as painful crises, and acute chest syndrome.
Acute painful episode
This refers to recurrent, severe painful episodes that can occur anywhere on the body, but typical sites include:
- Back
- Chest
- Abdomen
- Extremities
- Dactylitis: inflammation of a digit (i.e. fingers or toes) may be common in children.
The pain experienced during these episodes is highly variable and most times can be managed at home. In severe cases, patients require admission to hospital for parental analgesia (e.g. subcutaneous morphine) as the pain cannot be controlled. Around one third of patients may have daily painful episodes.
There are standardised protocols for managing patients with acute painful episodes, but often patients will have a personalised protocol. Importantly, acute painful episodes may occur concurrently with other life-threatening complications such as acute chest syndrome.
Acute chest syndrome
This describes a life-threatening syndrome, which is characterised by:
- Fever
- Chest pain
- Hypoxaemia
- Wheezing
- Cough
- Respiratory distress
On chest x-ray there is usually evidence of infiltration and patients may need urgent treatment to prevent serious complications. The cause of acute chest syndrome is multi-factorial and likely related concurrent infection, vaso-occlusion, hypoventilation and thrombosis.
Chronic complications
SCD is a multi-system disorder that can lead to profound organ damage.
- Neurological: cerebrovascular disease with deficits following stroke and seizure disorders.
- Blood and bone: chronic anaemia, osteoporosis and avascular necrosis.
- Cardiac: cardiomyopathy and heart failure (usually due to heart failure with preserved ejection fraction).
- Pulmonary disease: can develop problems with pulmonary hypertension
- Kidneys: chronic kidney disease
- Hepatobiliary: variety of mechanisms. Often damage to the liver from transfusional iron overload. Increased risk of pigment gallstones due to chronic haemolysis.
- Others: chronic pain, delayed puberty, leg ulcers and retinal disease
Diagnosis
SCD is diagnosed based on identification of haemoglobin S.
The majority of patients are diagnosed as part of the newborn screening programme within the UK. This aims to detect patients with SCD at birth. Babies at risk of SCD are usually identified from antenatal screening. This screens mothers, and if needed biological fathers, for abnormal haemoglobin variants (see below).
Newborn screening
This involves a blood spot sample (typically heel prick) that is taken on day 5 of life. The sample is then tested for nine rare, but serious conditions. These conditions include sickle cell disease, cystic fibrosis, congenital hypothyroidism and a series of inherited metabolic disorders.
Identification of newborns with sickle cell disease helps to reduce infant mortality and morbidity.
Laboratory techniques
There are several laboratory techniques that can be used to identify the abnormal sickle haemoglobin. Some techniques are discussed below:
- Haemoglobin electrophoresis: enables identification of the type of haemoglobin. Traditional method that involves movement of haemoglobin proteins in an electric field. This separates haemoglobin types into bands.
- High performance liquid chromatography (HPLC): technique that separates components within a mixture. Modern technique for detection of sickle haemoglobin. Results shown as different peaks.
- Capillary electrophoresis: similar to traditional haemoglobin electrophoresis. Haemoglobin types are separated using ion migration and electro-osmotic flow.
- DNA sequencing: allows direct identification of the beta globin gene sequence. Not typically required.
Other investigations
Patients with SCD typically have chronic anaemia (70-90 g/L), an elevated reticulocyte count due to haemolysis and evidence of sickle shaped erythrocytes on blood film microscopy. Lactate dehydrogenase is typically elevated and haptoglobin, which binds to free haemoglobin in plasma, is usually low suggesting haemolysis. Other investigations are guided by the suspected complication.
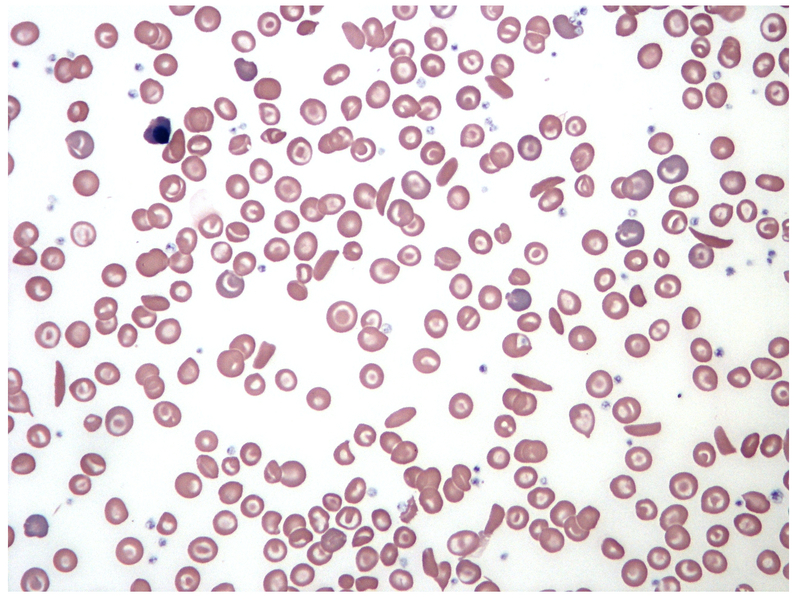
Blood film showing typical sickle-shaped erythrocytes
Image courtesy of Wikimedia Commons
Sickle solubility test
A sickle solubility test can be completed. Addition of a reducing solution to a blood sample causes sickling and the sample to become turbid. However, it is unable to differentiate between sickle cell anaemia and sickle cell trait. Additionally, other sickling haemoglobin variants will be positive.
Antenatal screening
Screening for sickle haemoglobin is offered to all women within the UK.
In the UK, women in high prevalence areas (>1.5 per 100,000 infants born with SCD) are offered antenatal screening at 10 weeks gestation. Screening determines if they are a carrier of sickle haemoglobin, other haemoglobin variants or thalassaemia. In low prevalence areas (<1.5 per 100,000 infants born with SCD), mothers are initially asked to fill in a Family Origin Questionnaire (FOQ). If mothers are found to have a sickle haemoglobin gene, the biological father is also offered screening to determine the risk of sickle cell anaemia (HbSS) or sickle cell disease (e.g. HbSC).
Depending on risk to the foetus, mothers may be offered the chance to undergo prenatal diagnosis (PND). PND involves determining an underlying problem early in pregnancy to enable decisions about continuation of pregnancy or planning for complications. The two major tests are chorionic villous sampling and amniocentesis. Newer cell free DNA tests are being explored for the SCD PND.
Management principles
Management involves patient education, treating acute and chronic complications and preventing vaso-occlusive phenomena.
The management of SCD is complex and should involve a haematologist who specialises in red cell disorders and there should be access to a multi-disciplinary team. A comprehensive overview of the management of SCD is far beyond the scope of these notes. Here we summarise the main management principles in SCD, which include:
- Patient education
- General care
- Managing acute complications (e.g. acute painful episodes)
- Preventing vaso-occlusive phenomena
- Stroke prevention
- Managing chronic complications
- Curative treatment
Patient education
Education is essential for patients, family members and carers. It involves education about the nature of SCD, pain management strategies, discussion about expected complications, infection prevention, preconception counselling and psychosocial involvement.
General care
All patients require regular follow-up with a haematologist who specialises in red cell disorders. This is generally 3-monthly in the first two years of life, 6-monthly up to the age of five, then annually as a minimum. Follow-up enables ongoing patient education and reinforcing key messages such as infection prevention and allows monitoring for chronic complications.
Due to chronic haemolysis and hyposplenism, all patients require folate supplementation and antibiotic prophylaxis.
- Folic acid replacement: at risk of bone marrow aplasia due to deficiency from increased red blood cell turnover. Usually 5mg daily.
- Antibiotic prophylaxis: usually penicillin prophylaxis (e.g. Pen V) or erythromycin in penicillin allergic patients
- Childhood immunisation: usual childhood immunisation programme with the addition of seasonal influenza, Pneumococcal (5-yearly from 2 years old), meningitis and hepatitis B vaccines.
- Adult immunisation: pneumococcal vaccine 5-yearly, seasonal influenza vaccine and further vaccinations (e.g. Meningococcal, hepatitis B, Haemophilus) depending on previous vaccinations.
Treating acute complications
Numerous acute complications occur in SCD. These are broadly divided into acute infections, anaemias (e.g. splenic sequestration, hyperhaemolysis) and vaso-occlusive phenomena. Many of these conditions have local guidelines to help manage patients appropriately. Detailed analysis of the management of all acute complications is beyond the scope of these notes.
The two most commonly encountered acute complications include:
- Acute painful episodes: also known as painful crisis or painful vaso-occlusive crisis.
- Acute chest syndrome: also known as chest crisis.
Both conditions are discussed in more detail in separate chapters below.
Preventing vaso-occlusive phenomena
There are a variety of treatment options used to reduce the frequency and severity of vaso-occlusive phenomena. These generally work by reducing the percentage of sickle haemoglobin (HbS) or increasing the foetal haemoglobin levels.
- Top-up transfusions: can be regular or ad hoc. Used to treat anaemia, manage complications (e.g. acute stroke, painful episode) or prepare for major surgery. Leads to reduction in HbS percentage. At risk of iron overload.
- Exchange transfusions: involves removal of a large proportion of the patients blood and replacing it with donor blood (i.e. 8 units). Can be given regularly as part of an exchange transfusion programme or used in emergencies to rapidly reduce the HbS percentage (i.e. acute chest syndrome). At risk of iron overload.
- Hydroxycarbamide/Hydroxyurea: cytotoxic oral medication that increases the level of foetal haemoglobin and reduces the amount of red cell sickling. Shown to reduce acute painful episodes and other acute complications (e.g. acute chest syndrome, stroke). Also decreases admission and prolongs survival. Various indications (discussed below).
The main indications for hydroxycarbamide/hydroxyurea include:
- ≥3 acute painful episode needing hospital admission
- Acute chest syndrome requiring transfusion or high-dependency unit admission
- Previous stroke (not on exchange transfusion programme)
New therapies
NICE has recently approved a new monoclonal antibody known as Crizanlizumab for the prevention of vaso-occlusive crises. It is an intravenous infusion that may be used as a stand alone therapy or alongside more traditional therapies such as hydroxycarbamide. Crizanlizumab targets P-selection, which is a cell adhesion molecule present on activated platelets and endothelial cells.
Stroke prevention
Stroke refers to neurological disturbances due to cerebral infarction (death of tissue from ischaemia). The management of stroke in SCD is usually with blood transfusions to reduce the HbS percentage. Reduction in sickle percentage is also the cornerstone of stroke prevention, which can be achieved with top-up transfusions for exchange transfusions.
In children, transcranial doppler ultrasound is performed annually between 2-16 years of age to determine the risk of stroke. Those at increased risk can be given regular blood transfusions. Children and adults who develop strokes can be offered the exchange transfusion programme as part of secondary prevention.
Managing chronic complications
Chronic complications may be due to the underlying disease process or from treatments (e.g. transfusions).
Recurrent sickling and vaso-occlusive phenomena leads to tissue ischaemia, infarction and chronic organ damage. Examples include cerebrovascular disease, cardiovascular disease and chronic kidney disease. Patients are monitored for long-term problems and established disease is managed by specialists.
One of the major problems with long-term transfusion treatment is iron overload. This can lead to secondary haemochromatosis with widespread organ dysfunction from iron deposition. Typical organs affected include the heart, liver, pancreas and pituitary gland. Patients may be given iron chelators (e.g. deferoxamine, deferasirox, and deferiprone) to prevent complications.
Curative treatment
Rarely, haematopoietic stem cell transplantation can be considered for patients, which would be curative. This is because the normal blood cells in the bone marrow are replaced with donor stem cells (e.g. matched sibling donor) that can differentiate into erythrocytes that contain haemoglobin without the beta globin gene mutation causing HbS.
Acute painful episodes
Acute painful episodes may be precipitated by hypoxia, infection, dehydration, cold weather or even pregnancy.
An acute painful episode, traditionally known as a ‘painful crisis’, can occur without warning and lead to severe pain due to vaso-occlusion within connective tissue (e.g. bone). They can occur anywhere on the body. Some painful episodes may be managed at home with simple analgesia, whereas others need inpatient admission for parental analgesia.
Investigations
Routine investigations are important to assess for other complications, any underlying precipitants and to assess haemoglobin and sickle percentage.
- Full blood count, reticulocyte count
- Group and save
- Biochemistry: LDH, U&Es, LFTs, Bone
- Septic screen: blood cultures (if fever), chest x-ray, mid-stream urine culture.
- Bone x-rays: rarely needed. Usually reserved for suspected acute fractures or osteomyelitis.
Management
Management of an acute painful episode is supportive and includes:
- Initial, rapid assessment (e.g. ABCDE)
- Observations: particular attention to saturations (to assess for evidence of acute chest syndrome).
- Check sickle care plan: personalised care plan for patients to manage painful episodes.
- Rapid administration of analgesia: usually subcutaneous morphine. Aim within 30 minutes of admission. Paracetamol/ibuprofen can be used as adjuncts.
- Regular assessment: regular observations, assessment of pain scores and administration of analgesia essential.
- Managing exacerbating factors: ensure well hydrated, treat concurrent infections and provide oxygen as needed (aim sats >94% unless chronic CO2 retention).
- Referral: all patients should be referred to their local haematology team.
- Transfusions: guided by the haematologists. Rarely needed in simple acute painful episodes.
Complications
Acute painful episodes can occur in conjunction with other acute complications (e.g. systemic infections, splenic sequestration, acute stroke). One of the major complications of an acute painful episode is acute chest syndrome. This should be suspected in any patient with increasing oxygen requirement, non-resolving pain, high fevers and/or infiltrates on the chest x-ray.
Acute chest syndrome
Around 50% of patients with SCD will have at least one episode of acute chest syndrome in their lifetime.
Definition
Acute chest syndrome can be a life-threatening condition. The cause is multi-factorial and can be very difficult to distinguish from other life-threatening pulmonary pathology (e.g. pulmonary embolism, adult respiratory distress syndrome).
Clinical features
It is characterised by fever, shortness of breath, tachycardia, hypoxia, chest pain and crackles on lung auscultation. On chest radiograph there is usually evidence of new pulmonary infiltrates that appear white. Acute chest syndrome may complicate patients who present with acute painful episodes.
Diagnosis & investigations
Acute chest syndrome is usually a clinical and radiological diagnosis based on characteristic clinical features in the context of new infiltrates on chest x-ray. Key investigations include:
- Bloods: FBC, U&E, Group & save, CRP, LFT
- Imaging: chest x-ray (looking for infiltrates)
- Infection screen: blood cultures, sputum cultures, viral PCR
- Arterial blood gas: ideally on room air to asses the degree of hypoxia.
Incentive spirometry
Incentive spirometry is a hand-held device that encourages deep breathing and aims to prevent atelectasis, which refers to incomplete lung expansion or partial collapse. Atelectasis causes ventilation/perfusion mismatch, worsens hypoxia and can precipitate acute chest syndrome.
Use is critical in patients with acute painful episodes at risk of acute chest syndrome and has been shown to reduce incidence.
Management
The principle management in severe acute chest syndrome with significant hypoxia is urgent exchange transfusion, which aims to rapidly reduce the sickle haemoglobin percentage. This can be automated or manual depending on time of day and staffing availability.
All patients require adequate hydration (i.e. IV fluids), antibiotics if concern regarding infection, adequate pain relief, physiotherapy (i.e. chest physio) and VTE prophylaxis if no contraindication.
Patients require frequent monitoring and early escalation to intensive care unit (ICU) if deterioration. Patients with severe hypoxia may require continuous positive airway pressure (CPAP) or invasive ventilation.
Prognosis
Sickle cell disease is a life-limiting condition, although modern medicine has greatly improved survival.
Overall survival differs between studies. Among children, >90% will survive beyond 20 years old. Older studies suggested a median survival of 42 years for men and 48 for women. However, a review of prognosis in a cohort of patients with sickle cell disease at King’s College Hospital showed a better median survival at 67 years old.
In developing countries with less robust preventative strategies infection is a major cause of death. In Developed countries, death is usually attributable to life-threatening acute complications (e.g. acute chest syndrome) or chronic organ failure (e.g. end-stage renal disease, pulmonary hypertension).
Last updated: October 2021
Have comments about these notes? Leave us feedback