Gilbert syndrome
Notes
Overview
Gilbert syndrome is an autosomal recessive disorder that causes abnormal bilirubin processing in the liver.
Gilbert syndrome is a benign, inherited cause of abnormal processing of bilirubin within the liver. This leads to recurrent episodes of unconjugated hyperbilirubinaemia.
Epidemiology
Gilbert syndrome is the most common cause of inherited jaundice.
Gilbert syndrome is seen in all populations and usually presents around the time of puberty. This is thought to be because sex hormones affect bilirubin processing in the liver. Gilbert is seen more commonly in males.
Bilirubin metabolism
Bilirubin is a breakdown product of heme metabolism.
Bilirubin is a breakdown product of haemoglobin, which is the oxygen-carrying pigment in red blood cells. Bilirubin is carried to the liver for processing where it can then be excreted into bile.
Formation of bilirubin
Red blood cells that contain haemoglobin are initially broken down by tissue macrophages, especially those located within the spleen. Heme is broken down by the enzyme heme oxygenase to form biliverdin, carbon monoxide and iron. Biliverdin is converted to bilirubin by biliverdin reductase.
Bilirubin then binds to albumin within the serum and is carried to the liver for processing. Bilirubin is then taken up by hepatocytes.
Bilirubin processing
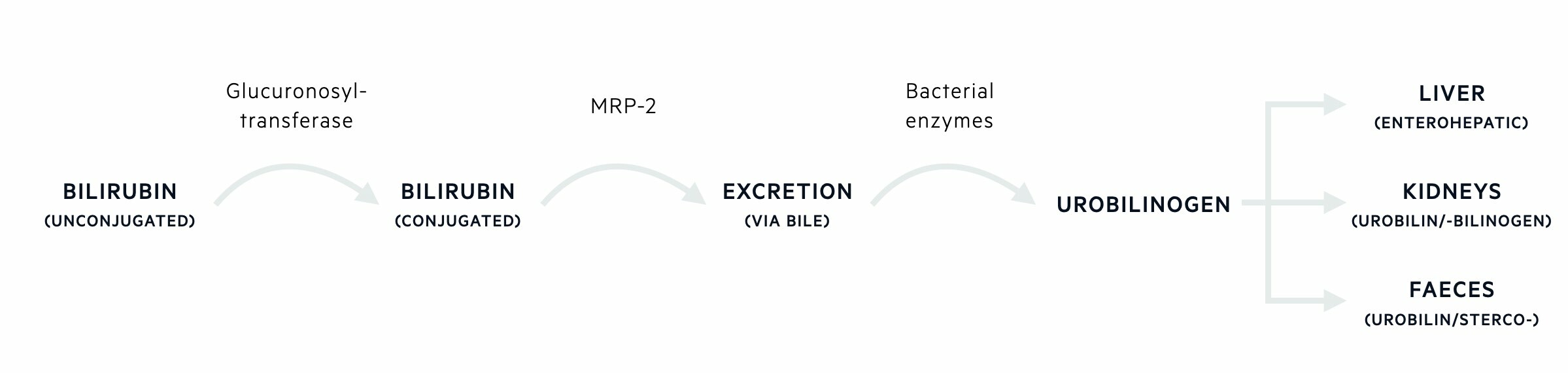
In the liver, bilirubin needs to undergo conjugation (addition of an extra molecule) to make it more water-soluble to allow it to be excreted into bile. This process of conjugation is known as glucuronidation because it involves the addition of glucuronic acid. The enzyme responsible for this is known as uridine diphosphoglucuronate-glucuronosyltransferase 1A1 (UGT1A1).
Bilirubin excretion
Once bilirubin has undergone glucuronidation, it is excreted into bile against its concentration gradient through bile canalicular membrane transporters such as multidrug resistance protein 2.
Bilirubin degradation
Bile is released into the gastrointestinal tract. Within the gastrointestinal tract, bilirubin is deconjugated by intestinal bacteria to a group of compounds known as urobilinogens. Urobilinogens may be further oxidised and some are reabsorbed via the enterohepatic circulation. These additional products include urobilin and sterocobilin, which are what give faeces its characteristic brown colour. A small percentage of the reabsorbed urobilinogens is excreted into urine. These oxidised urobilinogens account for the colored compounds that contribute to the yellow color of urine.
Aetiology & pathophysiology
Gilbert syndrome is due to impaired bilirubin glucuronidation.
Gilbert syndrome is an autosomal recessive disorder due to a genetic defect in the promoter region of the gene that encodes the enzyme uridine diphosphoglucuronate-glucuronosyltransferase 1A1 (UGT1A1). This enzyme conjugates (i.e. joins) bilirubin and glucuronic acid making it water-soluble and able to be excreted into bile.
The mutation in the promoter region leads to reduced activity of the enzyme (~30% of normal). This leads to recurrent unconjugated hyperbilirubinaemia with episodes of jaundice that are usually precipitated by certain ’stressful events’. Examples of precipitating events include:
- Fasting
- Haemolysis
- Febrile illness
- Physical exertion
- Stress
- Menses
Crigler-Najjar syndrome
Crigler-Najjar is an autosomal recessive condition due to several mutations in the same gene that is affected in Gilbert’s. Crigler-Najjar is more severe and usually presents in the neonatal period because it leads to the complete absence of enzymes activity (type 1) or enzyme activity that is < 10% (type 2).
Due to the severity of the mutation, it can lead to profound unconjugated hyperbilirubinaemia and subsequent neurological damage known as kernicterus. A series of life-long treatments are often required including daily phototherapy or phenobarbital.
Clinical features
Gilbert syndrome is characterised by recurrent episodes of jaundice.
Patients with Gilbert’s are usually asymptomatic until a stressful event or intercurrent illness leads to jaundice (typically noticed in the sclera). During these intermittent episodes of jaundice, patients may have non-specific symptoms including fatigue or abdominal discomfort.
Diagnosis & investigations
Diagnosis is based on recurrent unconjugated hyperbilirubinaemia and the absence of obvious liver disease or haemolysis.
A formal diagnosis of Gilbert syndrome does not require extensive testing. It can usually be made based on the presence of recurrent episodes of isolated unconjugated hyperbilirubinaemia and the absence of obvious haemolysis or liver disease.
Blood tests
Liver function tests (LFTs) will show an isolated rise in unconjugated bilirubin that should be confirmed on repeat testing. Total bilirubin is usually given as part of LFTs so it is important to ask for a ‘split bilirubin’ that gives both the conjugated and unconjugated portions. Other liver function tests (e.g. ALP, ALT) will be normal.
It is important to exclude haemolysis as a potential cause of a raised unconjugated bilirubin. This involves initial screening with a full blood count +/- a reticulocyte count and peripheral blood film. If the patient is anaemic or there are any signs of haemolysis then a full haemolytic screen should be requested.
Imaging
Ultrasound is usually not required to make a diagnosis of Gilbert’s if the history and blood results are consistent. A liver ultrasound will be normal.
Genetic testing
Genetic testing for specific mutations in the promoter region of the gene for UGT1A1 can be completed but is rarely necessary.
Management
No specific management is required for patients with Gilbert syndrome.
Gilbert syndrome is a benign condition without long-term sequelae. No specific treatment is indicated other than patient education.
Last updated: July 2022
Have comments about these notes? Leave us feedback