Wilson's disease
Notes
Overview
Wilson’s disease is a rare condition characterised by abnormal copper deposition.
Wilson’s is an inherited, multi-system, progressive disorder of copper metabolism. Named after British neurologist Dr Samuel Alexander Kinnier Wilson, it has an estimated incidence between 1 in 30,000 and 1 in 100,000.
It often presents with liver involvement or neuropsychiatric symptoms, but it can present in a number of other ways including haemolysis. Though it can develop at any age, it most commonly occurs between the ages of 5 and 35.
Aetiology
Wilson’s disease is an autosomal recessive disorder.
It is caused by mutations to the ATP7B gene on chromosome 13. About 500 different mutations causing Wilson's disease have been identified in this gene. ATP7B encodes a copper-transporting P-type ATPase that is found in hepatocytes (in the trans-Golgi network). The majority of these mutations are missense (single base pair change) that lead to loss-of-function. This means complete disruption of the normal copper-transporting protein function and the accumulation of copper in affected tissues.
As an autosomal recessive disorder, an offspring must receive an affected gene from both their mother and their father. When two carriers of a recessive allele have a child, there is a 25% chance of developing the condition, 50% chance of being a carrier (unaffected carrier) and a 25% chance of being entirely unaffected (non-carrier).
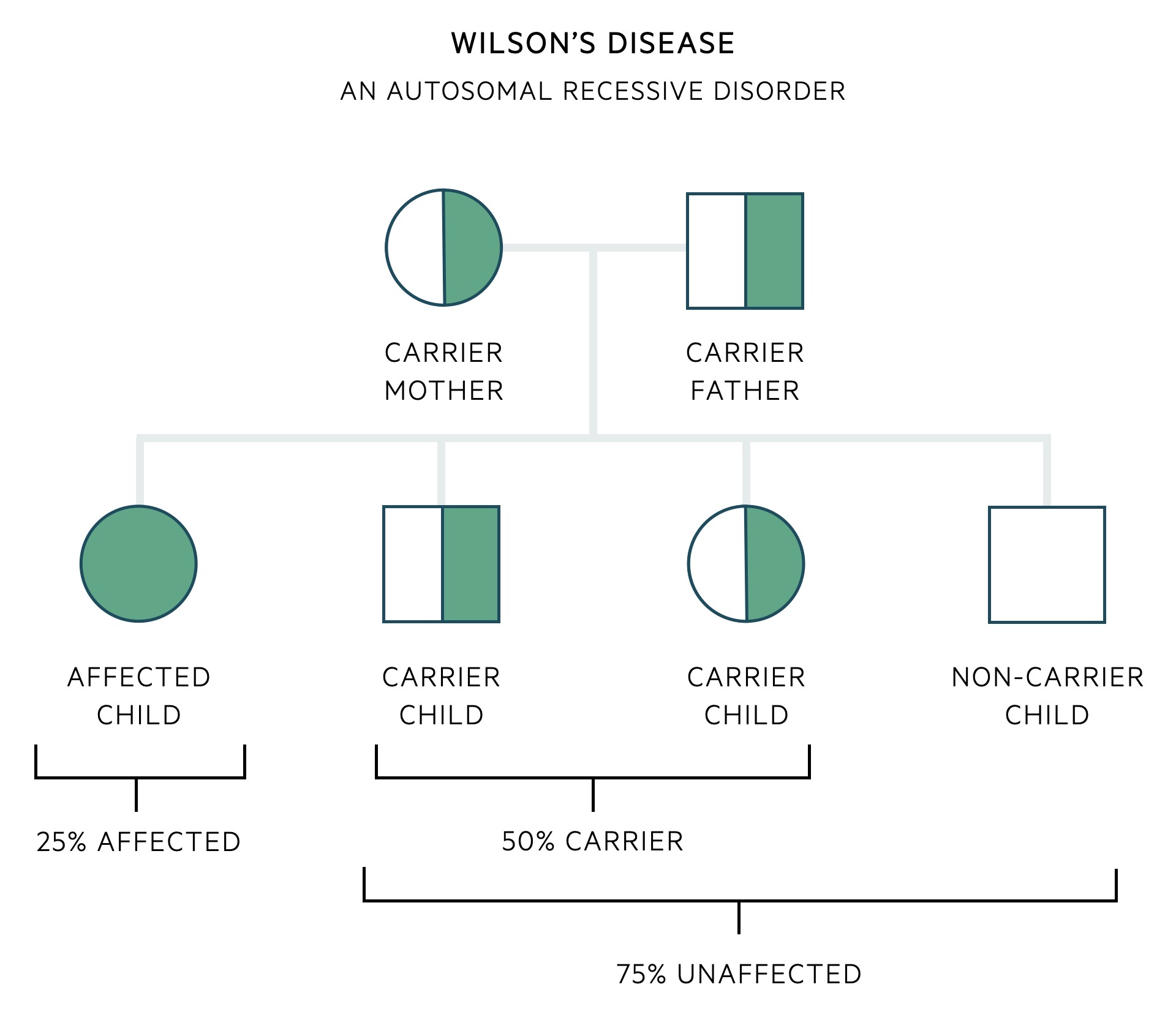
There are a multitude of mutations of the ATP7B gene that can lead to Wilson's disease. As such it is common for an affected patient to have inherited two different mutations, one from their mother and one from their father - these are referred to as compound heterozygotes. A compound heterozygote describes the inheritance of two different recessive alleles for the same gene, one on each chromosome (as opposed to homozygous if they had inherited the same mutation from each parent).
Clinical features
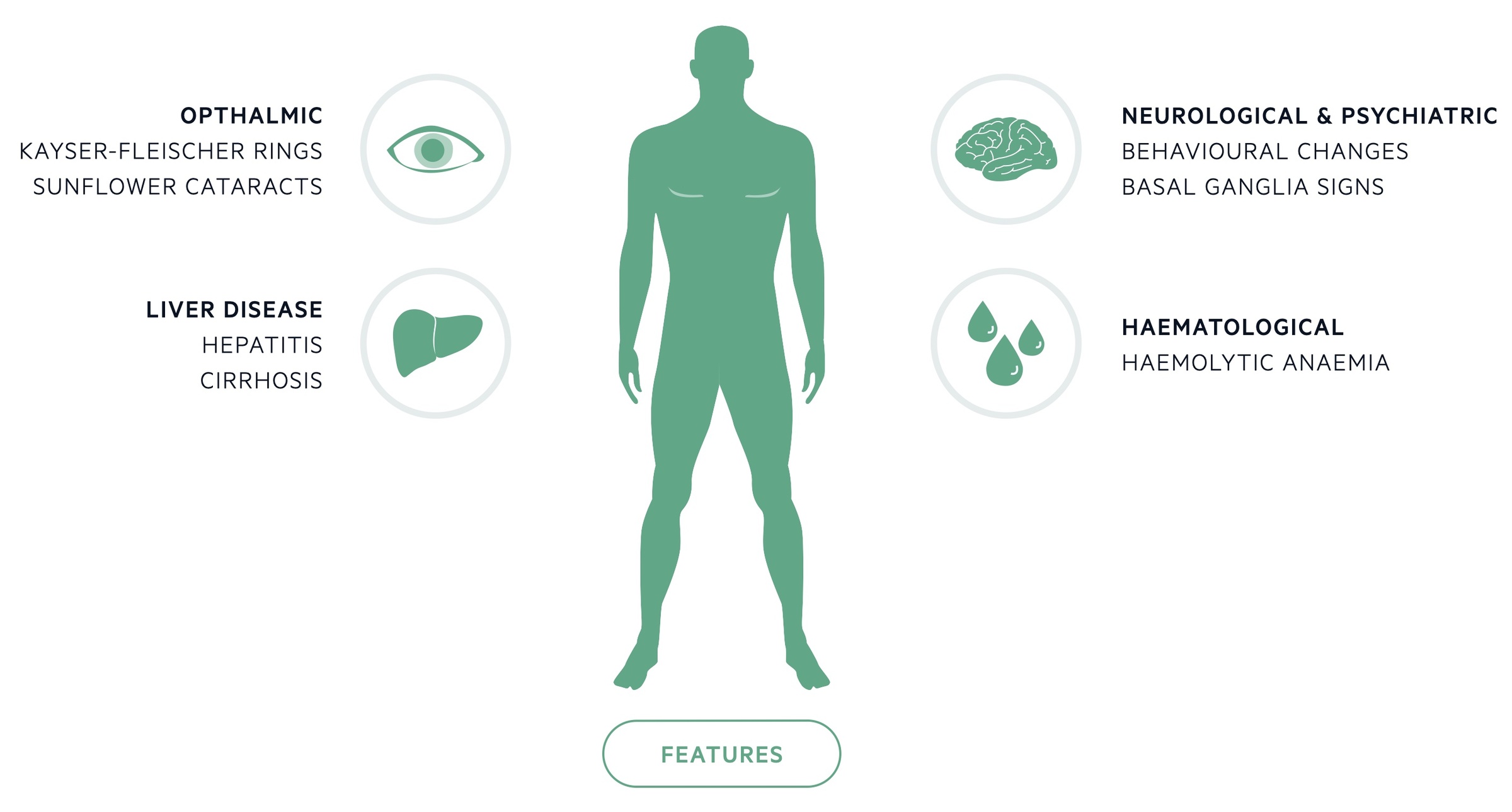
Wilson's disease most commonly presents due to hepatic or neurological involvement.
Ophthalmic
- Kayser-Fleischer rings: are copper deposits in Descemet’s membrane seen with slit-lamp examination. They are present in up to 95% of patients with symptomatic neurological disease but are far less common in those without neurological symptoms.
- Sunflower cataracts: are caused by copper deposits affecting the lens, they may be diagnosed with slit-lamp examination.
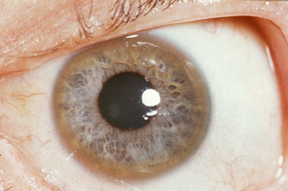
Kayser-Fleischer ring
Image courtesy of Wikipedia Commons
Liver disease
- Asymptomatic disease (with abnormal biochemistry)
- Acute liver failure
- Chronic hepatitis and cirrhosis
Neurological
- Akinetic-rigid syndrome: slow initiation of movement (similar to Parkinson's disease)
- Pseudosclerosis: clinical picture dominated by tremor, which is coarse and flapping
- Ataxia: both cerebellar ataxia and acroataxia (affecting the distal portion of limbs)
- Dystonic syndrome: slow, repetitive, involuntary muscle contractions
Psychiatric
- Behavioural changes (particularly younger patients)
- Lability of mood
- Impulsiveness
- Sexual exhibitionism
- Decline in academic performance
- Psychotic features
- Depression
Haematology
- Coombs-negative haemolytic anaemia (may be presenting feature)
Other
- Renal: renal tubular acidosis
- Rheumatology: osteoarthritis, myopathy, chondrocalcinosis
- Obstetric: miscarriages, infertility
Investigations
The diagnosis of Wilson’s disease relies on both clinical and biochemical findings.
Basic investigations
- Serum caeruloplasmin: caeruloplasmin is an acute phase reactant and the main carrier of copper in the blood. It tends to be decreased in patients with Wilson’s.
- Serum copper: tends to be decreased in line with serum caeruloplasmin, though it may be normal or elevated in acute liver failure. Serum ‘free’ copper (i.e. not bound to serum caeruloplasmin) tends to be elevated.
- 24-hour urinary copper: may aid the diagnosis of Wilson’s disease. It reflects the unbound serum ‘free’ copper. Tends to be elevated.
- Slit-lamp examination: Kayser-Fleischer rings, which are copper deposits in Descemet’s membrane, can be seen.
Further investigations
- Liver biopsy: an invasive investigation that is not without complications. Required if non-invasive tests are not diagnostic or to rule out other conditions.
- MRI/CT head: may reveal changes in the basal ganglia. The characteristic ‘face of the giant panda’ sign is seen in a minority of cases. See this Radiopaedia article for more on the giant panda sign.
- Genetic testing: should be offered, aids family screening. Challenging due to the hundreds of possible mutations and the fact that most are compound heterozygotes (carry two different mutations, see the aetiology section for more).
Family screening
Identification of at-risk relatives can prevent significant morbidity and mortality.
Genetic counselling should be offered with clear explanations about the condition and the mechanism of inheritance.
- Siblings: the sibling of a patient has a 25% risk of suffering from the disease. Analysis of the ATP7B gene should be carried out.
- Offspring: children of an affected patient should have analysis of the ATP7B gene.
Management
Treatment aims to reduce levels of copper and prevent further end-organ damage.
General
- Counselling: patients require counselling once the diagnosis has been made. The genetic nature of the condition should be explained.
- Lifestyle: advise a healthy lifestyle, avoiding potentially toxic drugs and alcohol.
- Follow-up: patients require lifelong follow-up and monitoring under a specialist unit.
Pharmacological
D-Penicillamine: the mainstay of Wilson’s management. It chelates copper and promotes its urinary excretion. It, unfortunately, has numerous side effects, many severe, which prohibits around one-third of patients from using it. Neurological symptoms can worsen at the start of treatment.
Trientine: also a chelation agent, trientine promotes urinary excretion of copper. It may be used as initial therapy or for those who do not tolerate D-penicillamine.
Liver transplant
Transplant may be required in patients presenting with acute liver failure or decompensated cirrhosis.
Orthotopic liver transplantation (removal of the patients liver which is replaced by a donor liver) results in normal hepatic handling of copper. Live, related (obligate heterozygote) transplantation offers excellent results.
Pregnancy
Prior to conception copper levels should be optimised. Haplotype analysis of their partner should be offered.
Treatment should continue in pregnancy but must be optimised and managed by a specialist. There is limited information on the safety of breast feeding whilst on chelation agents.
Prognosis
Wilson's is a progressive disease, early recognition and treatment is essential.
Untreated Wilson’s disease is fatal. The majority of patients die due to liver complications, though a minority will die from neurological complications.
Prognosis is dependent, in part, on time of diagnosis, with those diagnosed and treated early having better long-term outcomes. Specialist treatment and adherence to therapy is key to improving survival.
Last updated: November 2021
Have comments about these notes? Leave us feedback