Alport's syndrome
Notes
Overview
Alport’s syndrome is a rare hereditary nephropathy occurring due to mutations to genes encoding type IV collagen.
Alport’s may demonstrate different inheritance patterns though it is most commonly X-linked due to mutations of COL4A5 on the X chromosome.
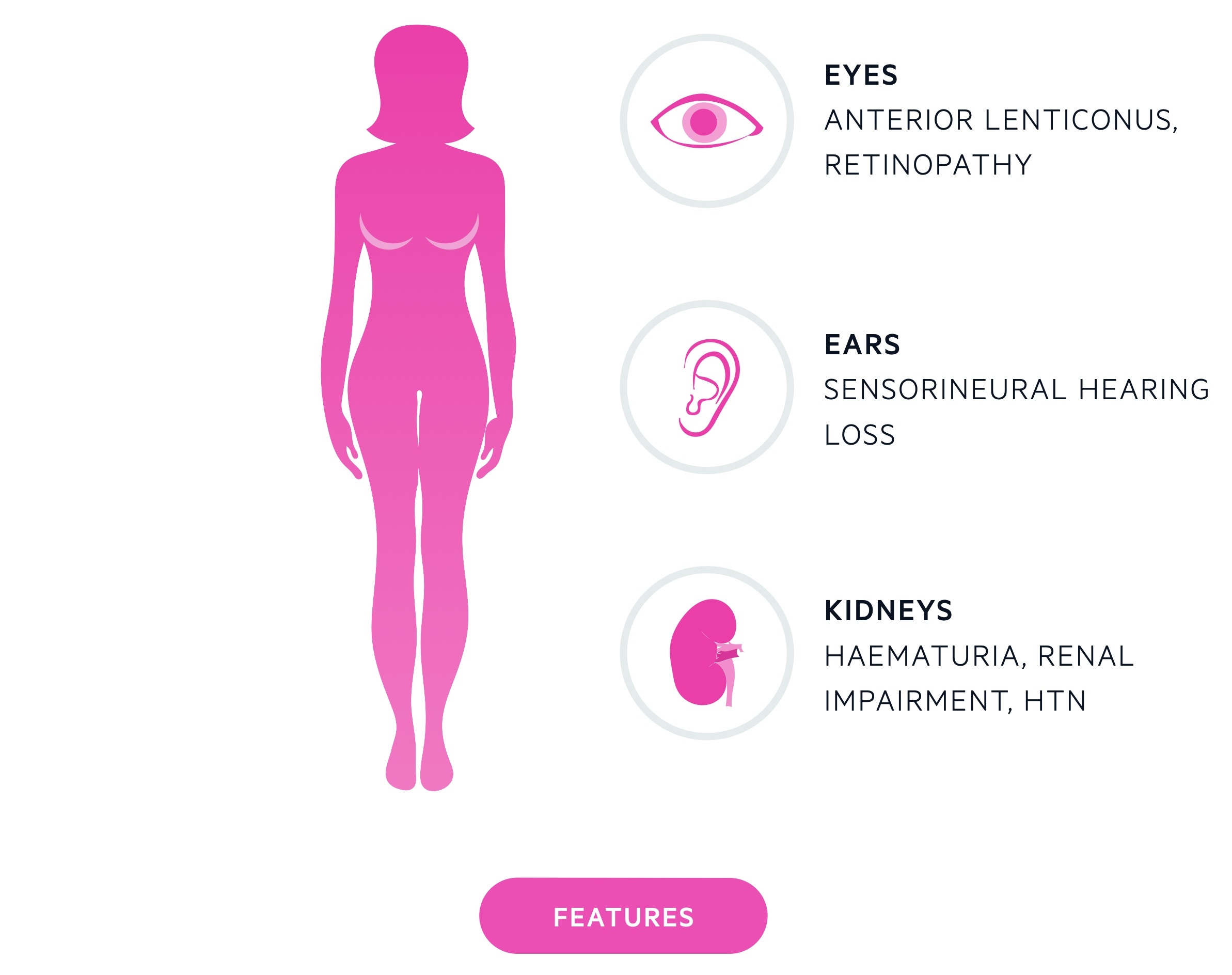
The disease affects the glomerular basement membrane (GBM) leading to nephritis. Other features may include sensorineural hearing loss and ocular defects.
Genetics
Alport’s may arise from a number of mutations to genes responsible for type IV collagen.
It may occur due to mutations to alpha-3, alpha-4 and alpha-5 chains of type IV collagen. Type IV collagen is important to basement membranes and mutations lead to impaired function. This affects basement membranes in the kidneys (GBM), eyes and ears.
Inheritance may be X-linked (most common), autosomal dominant or autosomal recessive. Rarely autosomal digenic inheritance is seen.
X-linked
The majority of patients are affected by the X-linked form. It occurs due to mutations of COL4A5. Female carriers (i.e. heterozygotes) of the X-linked form are not unaffected due to lyonisation, they have an increased risk of hypertension and renal impairment. Up to 20% will be affected by sensorineural hearing loss.
NOTE: Lyonisation refers to the process of inactivation of one of the X chromosomes on a cellular level in females.
Autosomal dominant
Accounts for around 20-30% of cases. Occurs due to mutations to COL4A3 or COL4A4. Typically has a mild phenotype.
Autosomal recessive
Accounts for around 15% of cases. Occurs due to mutations to COL4A3 or COL4A4, may be associated with parental consanguinity.
Clinical features
Alport’s syndrome leads to microscopic haematuria, progressive renal failure and hypertension.
Renal effects
The first identifiable sign is asymptomatic microscopic haematuria that manifests in childhood. Unless specifically screened for or episodes of macroscopic haematuria occur this is unlikely to be picked up.
As affected individuals grow up renal impairment and proteinuria develop. Renal impairment causes a secondary hypertension.
Hearing loss
Bilateral sensorineural hearing loss, initially affecting high frequencies, is often seen.
Ocular effects
There are a number of ocular pathologies seen in patients with Alport’s:
- Anterior lenticonus: refers to protrusion of the lens due to weakness of the lens capsule. Pathognomonic but only seen in around 20-30% with the X-linked form.
- Retinal granulations: white / yellow granulations seen on the retina.
- Cornea: multiple changes may occur including recurrent corneal erosions.
Other
Patients may develop leiomyomas and arterial aneurysms amongst other manifestations.
Diagnosis
The diagnosis of Alport’s can be confirmed with genetic testing or biopsy (of the skin or kidneys).
Genetic testing
Molecular genetic testing can confirm the diagnosis of Alport’s syndrome and the underlying mutation type.
Biopsy
There are two tissue types that may be biopsied:
- Skin: has the advantage of being less invasive but is primarily of use for suspected X-linked disease.
- Kidney: review of the GBM can show changes characteristic of Alport's syndrome.
Management
Kidney transplant offers a chance of ‘cure’ in patients with Alport’s.
General management
Patients require lifelong care under a nephrologist. Regular monitoring of renal function, proteinuria and blood pressure is required amongst other investigations.
In patients with hypertension and proteinuria, ACE inhibitors are the preferred initial therapy. Genetic counselling should be offered to all patients (including prior to genetic testing).
Kidney transplant
Transplant offers a chance at a ‘cure’ in patients with chronic kidney disease. Patients typically respond well and it alleviates the need for dialysis.
Though Alport’s cannot recur in the transplant kidney, approximately 1-5% will develop anti-GBM disease.
Extra-renal disease
Patients with sensorineural hearing loss or ocular pathology should be referred to audiology/ENT and ophthalmology. Interventions are typically supportive and may include hearing aids.
Patients with leiomyomas will occasionally require surgical resection.
Last updated: June 2021
Have comments about these notes? Leave us feedback