Ventilation
Notes
Ventilation
Ventilation is the movement of air between the lungs and the surrounding environment.
Ventilation is key to maintaining adequate arterial oxygenation and for the removal of CO2. Under normal circumstances, breathing is a passive process controlled by centres in the brainstem. These respiratory centres are specialised groups of neurones. The complex interaction of these neuronal collections are responsible for ventilation.
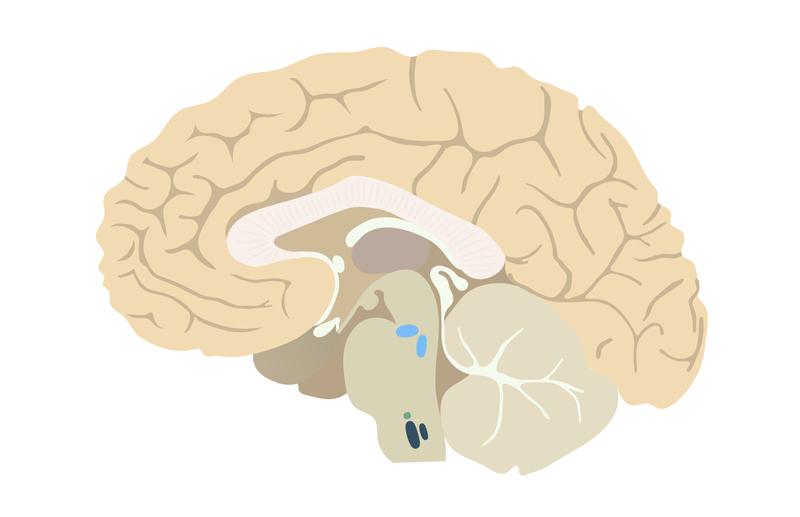
Three such groups have been identified: the pons respiratory centre, the medullary respiratory centre and the pre-Bötzinger complex.
Respiratory centres
Pons respiratory centre
The pons (primary) respiratory centre is composed of two centres.
- Pneumotaxic centres: can interact with the dorsal respiratory group (see below) to suppress inspiration.
- Apneustic centres: acts to stimulate inspiration.
Medullary respiratory centre
The medullary respiratory centre is composed of two groups.
- Dorsal respiratory group (DRG): composed of inspiratory neurones.
- Ventral respiratory group (VRG): composed of inspiratory and expiratory neurones. Utilised in times of increased ventilatory needs.
Pre-Bötzinger complex
Respiratory rhythm is thought to be generated by the pre-Bötzinger complex. The neurones here exhibit pacemaker-like activity which is thought to drive the DRG.
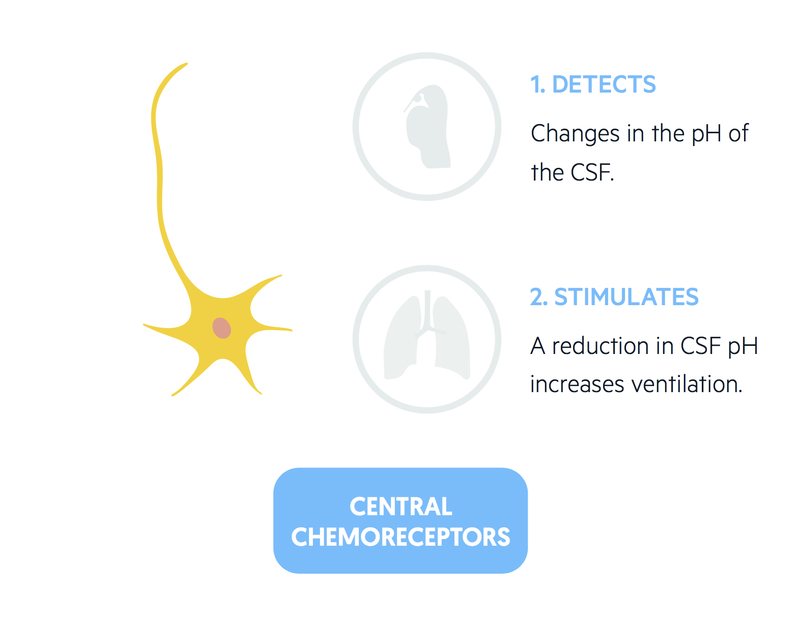
Central chemoreceptors
Central chemoreceptors exert significant influence over the respiratory centres in response to changes to acid/base balance.
Located on the ventrolateral surface of the medulla, central chemoreceptors are sensitive to changes in pH within the cerebrospinal fluid (CSF). The central nervous system itself is separated from the blood by the blood-brain barrier which hydrogen ions (H+) are unable to cross.
Instead, carbon dioxide (CO2) levels are responsible for pH changes in the CSF. CO2, which is lipid-soluble, easily diffuses through the blood-brain barrier. In the CSF, it forms carbonic acid with water. Carbonic acid then may dissociate to hydrogen ions and bicarbonate. An increase in hydrogen ions leads to a reduction in the CSF pH.
- A reduction in CSF pH (caused by an increase in CO2) causes an increase in ventilation.
- An increase in CSF pH (caused by a reduction in CO2) causes a reduction in ventilation.
Respiratory acidosis - caused by raised CO2 - is quickly detected via the mechanism described above leading to compensatory changes. The impact of systemic metabolic acidosis on this system is not fully understood.
It is thought that a systemic metabolic acidosis like diabetic ketoacidosis or lactic acidosis stimulates central chemoreceptors. Animal studies have shown activation of central chemoreceptors in metabolic acidosis may occur, likely due to subtle changes in CSF pH.
Local cerebral hypoxaemia will also activate central chemoreceptors due to the development of a localised metabolic acidosis.
This system, though slower than the peripheral chemoreceptors, is the primary stimulus of ventilation and is estimated to provide 80% of the stimulus for a ventilatory response to a raised arterial CO2.
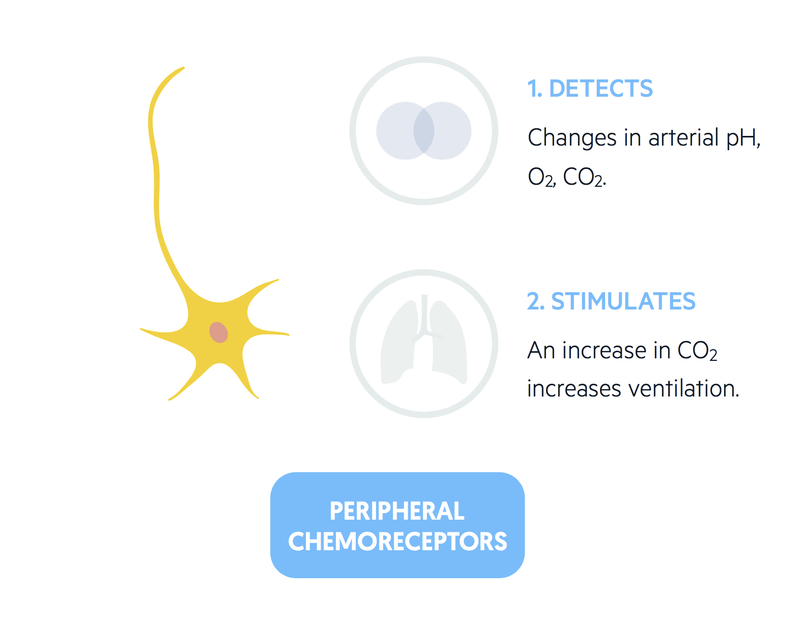
Peripheral chemoreceptors
Peripheral chemoreceptors are able to bring about rapid changes to ventilation in response to fluctuations in arterial pH, O2 and CO2.
There are two sets of peripheral chemoreceptors:
- Carotid bodies: found near the bifurcation of the common carotid. Capable of responding to arterial pH, O2 and CO2.
- Aortic bodies: found in the aortic arch. Capable of responding to arterial O2 and CO2.
These ‘bodies’ are bathed in blood thanks to sinusoidal capillaries. A reduction in O2 or an increase in CO2 causes an increase in ventilation.
They act more rapidly than central chemoreceptors and are estimated to provide 20% of the stimulus for a ventilatory response to raised arterial CO2.
Last updated: April 2021
Have comments about these notes? Leave us feedback