Familial hyperlipidaemia
Notes
Overview
Familial hyperlipidaemia refers to the inheritance of a single (or multiple) genetic variant(s) that lead to elevated lipid levels.
Familial hyperlipidaemia is also known as primary hyperlipidaemia. It refers to the inheritance of single or multiple genetic variants that cause an abnormal elevation in serum lipids. These lipids include total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG). If there is a single genetic variant (i.e. mutation) this is known as a monogenic disease. If there are multiple genetic variants that all contribute to the development of the condition this is known as a polygenic disease.
The pattern of lipid abnormalities (e.g. elevated low-density lipoprotein cholesterol or elevated triglycerides) depends on which gene is abnormal and what role it has in lipid metabolism. There are many different familial hyperlipidaemias that each have a complex underlying genetic basis. The principal problem with familial hyperlipidaemia is early-onset cardiovascular disease that can be significantly life-limiting in some cases.
For more information on non-familial hyperlipidaemia, check out our Hyperlipidaemia notes.
Classification
The classification of familial hyperlipidaemia is complex and very heterogeneous with many contributing genetic factors.
The traditional classification of lipid disorders was based on the Fredrickson classification in the 1960-70s, which was later adopted by the World Health Organisation (WHO). This classification grouped primary hyperlipidaemias into types I-V based on the pattern of abnormal lipid levels due to disturbances in underlying lipoprotein metabolism.
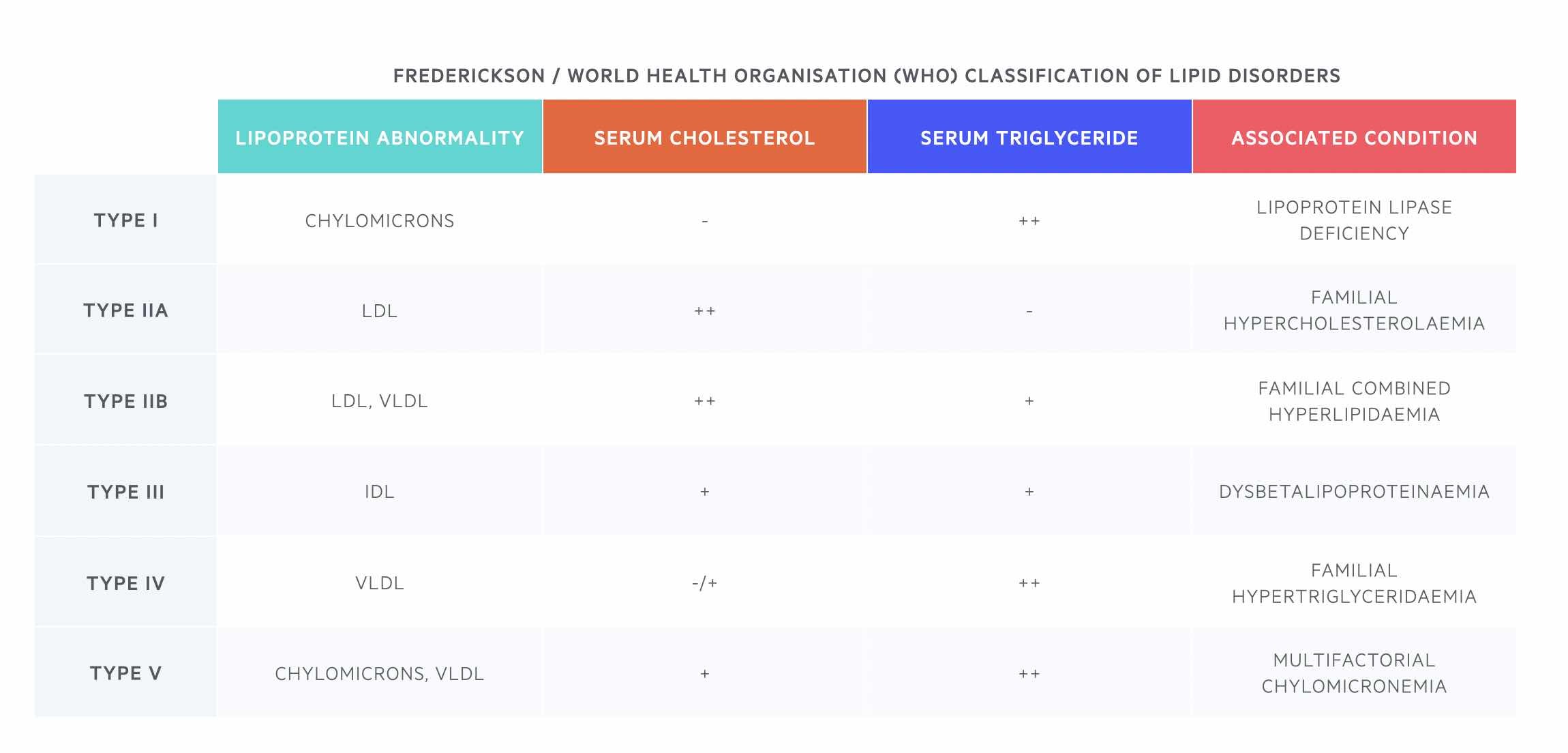
In this classification system, each type can refer to one or more clinical disorders (e.g. type II may be due to familial hypercholesterolaemia). This classification system has several shortcomings and is no longer used routinely in clinical practice. Various categorisations of primary lipid disorders are now based on pathological findings and genetic abnormalities.
Referral (suspected familial)
Patients with, or suspected to have, primary hyperlipidaemia should be referred to a specialist lipid clinic.
When determining a patients' risk of familial hyperlipidaemia it is important to consider the serum lipid levels, clinical features (e.g. any xanthelasma or xanthoma) and family history of lipid disorders or early cardiovascular disease.
The national institute of clinical excellence (NICE) produced guidelines for Familial hypercholesterolaemia: identification and management (CG71) in 2008 with updates in 2019. These coincide with their guidelines on Cardiovascular disease: risk assessment and reduction, including lipid modification (CG181) produced in 2008 with updates in 2016.
Suspecting familial hypercholesterolaemia
Familial hypercholesterolaemia should be suspected if:
- Total cholesterol > 7.5 mmol/L, AND/OR
- Personal or family history (first-degree relative) of premature coronary artery disease (a cardiovascular event < 60 years)
In those with a personal or family history of premature coronary artery disease, a serum lipid profile should be offered if not already completed. The Simon Broome criteria or Dutch Lipid Clinic Network criteria should then be used in those with suspected familial hypercholesterolaemia to make a clinical diagnosis (see below). These patients should be referred to a specialist lipid clinic.
Specialist referral
A specialist referral to a lipid clinic should be made regardless of family history if:
- Clinical diagnosis of FH, OR
- Total cholesterol > 9.0 mmol/L, AND/OR
- LDL cholesterol > 6.5 mmol/L, AND/OR
- Non-HDL cholesterol > 7.5 mmol/L OR
- Fasting triglycerides > 10 mmol/L
An urgent specialist review should be arranged for those with a TG concentration > 20 mmol/L in the absence of excess alcohol or poor glycemic control. The finding of significantly elevated cholesterol and/or triglycerides helps to identify patients likely of having underlying primary hyperlipidaemia.
Simon Broome Criteria
These criteria were developed in the 1980s in the UK. It can be used to make a ‘definitive’ or ‘probable’ diagnosis of familial hypercholesterolaemia. These patients should be referred to a specialist lipid clinic.
Definitive criteria
Total cholesterol > 6.7 mmol/L or LDL-C > 4.0 mmol/L in a child < 16 years old, OR total cholesterol > 7.5 mmol/L or LDL-C > 4.9 mmol/L in an adult, WITH
- Tendon xanthomas in patient or 1st/2nd-degree relative, OR
- Evidence of genetic mutation in LDL receptor or defective Apo B-100 or PCSK9 mutation
Probable criteria
Total cholesterol > 6.7 mmol/L or LDL-C > 4.0 mmol/L in a child < 16 years old, OR total cholesterol > 7.5 mmol/L or LDL-C > 4.9 mmol/L in an adult, WITH
- Family history of myocardial infarction (< 50 years 2nd-degree relative or < 60 years in a 1st-degree relative), OR
- Family history of TC > 7.5mmol/L in adult 1st/2nd degree relative, OR > 6.7mmol/L in child/sibling < 16 years
Familial hypercholesterolaemia
Familial hypercholesterolaemia is a common inherited disorder of LDL lipid metabolism.
Familial hypercholesterolaemia (FH) is a common cause of primary hyperlipidaemia that is due to genetic defects in low-density lipoprotein (LDL) metabolism. It is characterised by significant elevations in TC and LDL-C with early-onset coronary artery disease (CAD).
It is considered a monogenic condition (i.e. a single genetic variant causes the phenotype) that is inherited in an autosomal dominant pattern. The prevalence of the heterozygous form (i.e. inheritance of a single abnormal gene) is estimated at 1 in 300-500 in the general population.
Aetiology
FH is one of the most common autosomal dominant genetic disorders. A mutation may be identified in one of three keys genes:
- Low-density lipoprotein receptor (LDLR) gene: also referred to as the ApoB/E receptor
- Proprotein convertase subtilisin kexin 9 (PCSK9) gene
- Apolipoprotein B gene
An LDLR mutation is the most common. Patients may have a single abnormal gene (heterozygous) or two abnormal genes (homozygous). Patients with homozygous FH develop severe, premature CAD and may die at a young age. Thankfully, homozygous FH is rare with a prevalence of 1 in 300,000 to 400,000.
Clinical features
FH should be suspected in patients with a significantly elevated LDL-C, personal or family history of early-onset CAD, sudden premature cardiac death, or evidence of cholesterol deposits in the skin.
Features frequently found in patients with FH include:
- Tendon xanthomata: hard, non-tender nodular enlargement of tendons (common on knuckles, Achilles, extensor surfaces)
- Skin xanthomata: raised, waxy-appearing, yellow plaques on the skin
- Xanthelasma: yellow plaques on the upper eyelids
Diagnosis & investigations
A formal diagnosis of FH can be made using genetic testing to look for a pathogenic mutation in one of the three commonly identified genes. Alternatively, the Simon Broome or Dutch Lipid Clinic Network criteria can be used that do not require formal genetic testing. The Simone Broome criterion is commonly used in the UK and groups patients into 'definite' and 'probable' heterozygous FH. The clinical criterion for homozygous FH is slightly different.
To enable the use of the Simon Broome criteria, it is important to take a full clinical history, perform a clinical examination and take a full serum lipid profile. Patients with suspected FH need a referral to a specialist lipid clinic.
Management
All patients should be advised on appropriate lifestyle modifications in addition to anti-lipid therapy. For more information on lifestyle modification see our Hyperlipidaemia note.
A range of anti-lipid therapies are used in the management of FH to reduce LDL-C as much as possible. A broad aim is to achieve at least a 50% reduction of LDL-C (or non-fasting non-HDL-C) from baseline.
Options include;
- High-intensity statin therapy
- Ezetimibe: alone in those unable to have statins or in combination
- PCSK9 inhibitors (e.g. alirocumab)
A variety of additional options are usually restricted to patients with homozygous FH and include:
- Evinacumab (new therapy for homozygous FH)
- Lomitapide (inhibits microsomal triglyceride transfer protein)
- Other therapies (e.g. aphesis, liver transplantation)
Polygenic hypercholesterolaemia
Polygenic hypercholesterolaemia causes significantly elevated LDL-C levels in the absence of a single pathogenic mutation.
Polygenic hypercholesterolaemia is also known as common hypercholesterolaemia. It is characterised by a significantly elevated LDL-C level. This increase the risk of premature CAD. The condition is similar to FH but it occurs without an identifiable single pathogenic genetic mutation. Instead, multiple genes (i.e. polygenic) individually contribute to a significantly elevated cholesterol level.
Aetiology
The is a very heterogeneous group of conditions. Some patients may have a small number of highly penetrant genes contributing to the development of hypercholesterolaemia, whereas others may have many low penetrant genes.
The genetics is poorly understood and thought to affect LDL metabolism at multiple levels.
Clinical features
Polygenic hypercholesterolaemia is characterised by high cholesterol levels (TC + LDL-C) and premature CAD. The elevation in cholesterol is usually less compared to FH. There is usually a strong family history but typical clinical features (e.g xanthomas) are usually absent.
Diagnosis & investigations
The diagnosis is usually based on the evidence of early CAD, strong family history, absence of clinical findings, and significantly elevated LDL-C. Genetic testing may be completed to exclude FH but routine testing is not completed as it has not been shown to improve clinical outcomes.
Management
All patients should be advised of lifestyle modifications and the principal treatment is with high-intensity statins. Additional anti-lipid agents can be used if intolerance develops or there is an inadequate fall in lipid levels. Options include ezetimibe and PCSK9 inhibitors.
Familial combined hyperlipidaemia
Familial combined hyperlipidaemia is characterised by elevated cholesterol and triglycerides.
Familial combined hyperlipidaemia is a common genetic lipid disorder that affects 1-2% of the general population. The condition is complex with multiple genes contributing to the development of the condition through their interaction with the environment. As the name suggests, it is characterised by elevations in both LDL-C and triglycerides.
Aetiology
Familial combined hyperlipidaemia is considered a polygenic disorder. This means there are many genes that contribute to developing the condition. In many cases, it is due to an overproduction of ApoB-100-containing lipoprotein particles (VLDL/LDL). The severity of raised LDL-C or TG varies between patients depending on the underlying genetic mutations and environmental factors.
Clinical features
The condition is characterised by premature CAD, xanthelasma and obesity. In those with markedly elevated triglycerides, diabetes mellitus is commonly observed.
Diagnosis & investigations
The diagnosis of familial combined hyperlipidaemia is suspected when there are significantly elevated levels of both LDL-C and triglycerides. It is important to exclude secondary causes (e.g. poorly controlled diabetes mellitus). However, given that the levels of LDL-C and triglycerides can vary, the condition is suggested by high concentrations of Apolipoprotein B. The measurement of ApoB helps to differentiate the condition from other disorders such as dysbetalipoproteinaemia.
Management
First-line treatment in familial combined hyperlipidaemia is statin therapy. The addition of ezetimibe should be considered if cholesterol treatment targets are not reached. In general, patients with significantly elevated triglycerides despite statin therapy should be managed by a lipid specialist.
Polygenic hypertriglyceridaemia
Polygenic hypertriglyceridaemia is also known as primary hypertriglyceridaemia.
There are multiple causes of hypertriglyceridaemia that can make it difficult to differentiate between causes.
In polygenic hypertriglyceridaemia, previously known as primary hypertriglyceridaemia, there are multiple genetic traits that increase the risk of high TG through increased production of VLDL. It is thought to show an autosomal dominant pattern of inheritance.
Aetiology
Polygenic hypertriglyceridaemia is one of the primary causes of hypertriglyceridaemia. It is due to multiple genetic traits that cause an increase in triglyceride-rich VLDL particles. It is characterised by high triglycerides but normal cholesterol and ApoB concentrations.
Clinical features
Patients with high TG levels have an increased risk of acute pancreatitis and there is an association with early-onset CAD. Some patients may develop xanthomas. Eruptive xanthoma is a classic presentation. These are crops of 2-5 mm yellow papules that commonly occur over extensor surfaces but can be widespread. They can be tender and itchy and may occur from high levels of triglycerides (> 11.2 mmol/L) of any cause.
Diagnosis & investigations
The diagnosis of hypertriglyceridaemia is made by the identification of elevated TG on a fasting serum lipid sample. In general, patients with fasting triglycerides > 10 mmol/L should be referred to a specialist lipid clinic and if > 20 mmol/L this should be urgent.
Familial hypertriglyceridaemia usually shows elevated triglycerides with normal LDL-C and low HDL-C levels.
Management
It is important to address modifiable risk factors for hypertriglyceridaemia such as diet, alcohol, and weight loss. In patients at risk of cardiovascular disease statin therapy can be used. In those with significantly elevated triglycerides despite lifestyle measures then additional drugs can be used including fibrates, omega-3 fatty acid and newer agents such as icosapent ethyl are being investigated.
Chylomicronaemia
Chylomicronaemia may be due to a single genetic variant (i.e. monogenic) or multiple genetic variants (i.e polygenic).
High levels of chylomicrons due to inherited genetic variants of lipid metabolism is complex.
Multifactorial chylomicronemia traditionally falls under type V on the Fredrickson/WHO classification. It has a complex genetic background and results in elevated chylomicrons and VLDL. The phenotype is usually bought out in the presence of other environmental (i.e. acquired) factors. Due to the high levels of chylomicrons and VLDLs, patients will have evidence of hypertriglyceridaemia.
Monogenic chylomicronemia traditionally falls under type I on the Fredrickson/WHO classification. It is characterised by severe hypertriglyceridaemia (> 10 mmol/L). It is a rare autosomal recessive disorder due to lipoprotein lipase deficiency. It causes significantly elevated levels of chylomicrons and triglycerides.
Last updated: June 2022
Have comments about these notes? Leave us feedback