Microcytic anaemia
Notes
Overview
Microcytic anaemia describes the presence of a reduced haemoglobin concentration and a reduction in the mean corpuscular volume (MCV).
Normal RBC haemoglobin (Hb) concentration for males is approximately 130-175 g/L and for females 120-155 g/L. Hb concentration below these levels is considered to be anaemic.
The MCV describes the mean volume of erythrocytes and is measured in femtolitres (fL). The standard range for erythrocytes is 82-99 fL. Levels < 82 fL are considered microcytic.

There are numerous causes of a microcytic anaemia:
- Iron-deficiency anaemia (IDA)
- Anaemia of chronic disease
- Thalassaemias (e.g. alpha / beta)
Iron-deficiency anaemia
IDA is the most common cause of anaemia worldwide.
Epidemiology
IDA is commonly seen in women of child-bearing age and children across the world. Premenopausal females are particularly at risk because of the loss of iron during menstruation and pregnancy.
In the developed world, it is estimated that 2-5% of adult men and postmenopausal women suffer from IDA.
Dietary deficiency of iron is uncommon in the developed world because of adequate access to dietary components such as meat. Where meat is not a core component of the diet, the prevalence of IDA is 6-8 times greater.
Aetiology & pathophysiology
Iron is an essential molecule for living organisms; it is vital for numerous cellular processes including oxygen transport & DNA synthesis.
The absorption of iron from enterocytes in the gastrointestinal tract is highly regulated to match the loss of iron from the body each day. When the rate of iron absorption cannot keep up with the rate of iron loss, it will lead to depletion of iron stores within the body and eventually IDA.
Each day, around 1-2 mg of iron is absorbed and 1-2 mg is lost from the body.
The total iron content within our body is approximately 3-4 grams, which is distributed among different structures:
- Hb: 2-3 grams
- Plasma iron (e.g. bound to transferrin): 3-7 mg
- Iron-containing proteins (e.g. myoglobin): 300-400 mg
- Stored iron (e.g. ferritin, haemosiderin): 1 gram
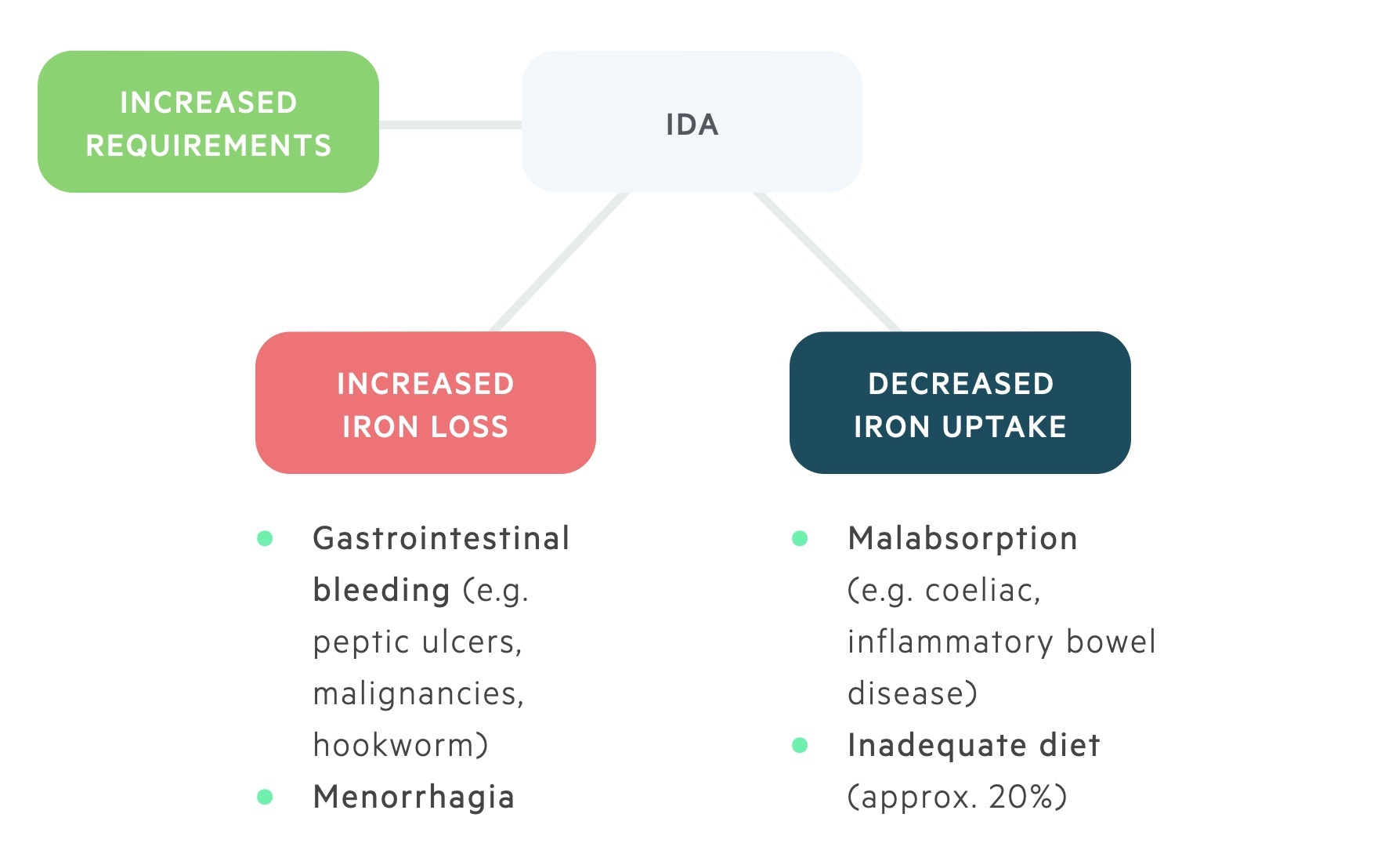
The major causes of IDA can be grouped into three categories:
- Increased requirements (e.g. pregnancy, lactation)
- Increased loss (e.g. gastrointestinal bleeding)
- Decreased uptake (e.g. dietary deficiency, malabsorption)
Chronic haemorrhage (e.g. increased iron loss) is one of the most common causes of IDA in the developed world. It is commonly due to excessive menstruation loss in premenopausal women but can be suggestive of underlying gastrointestinal pathology.
Clinical features
Clinical features reflect a reduction in Hb concentration and an impairment in the oxygen-carrying ability.
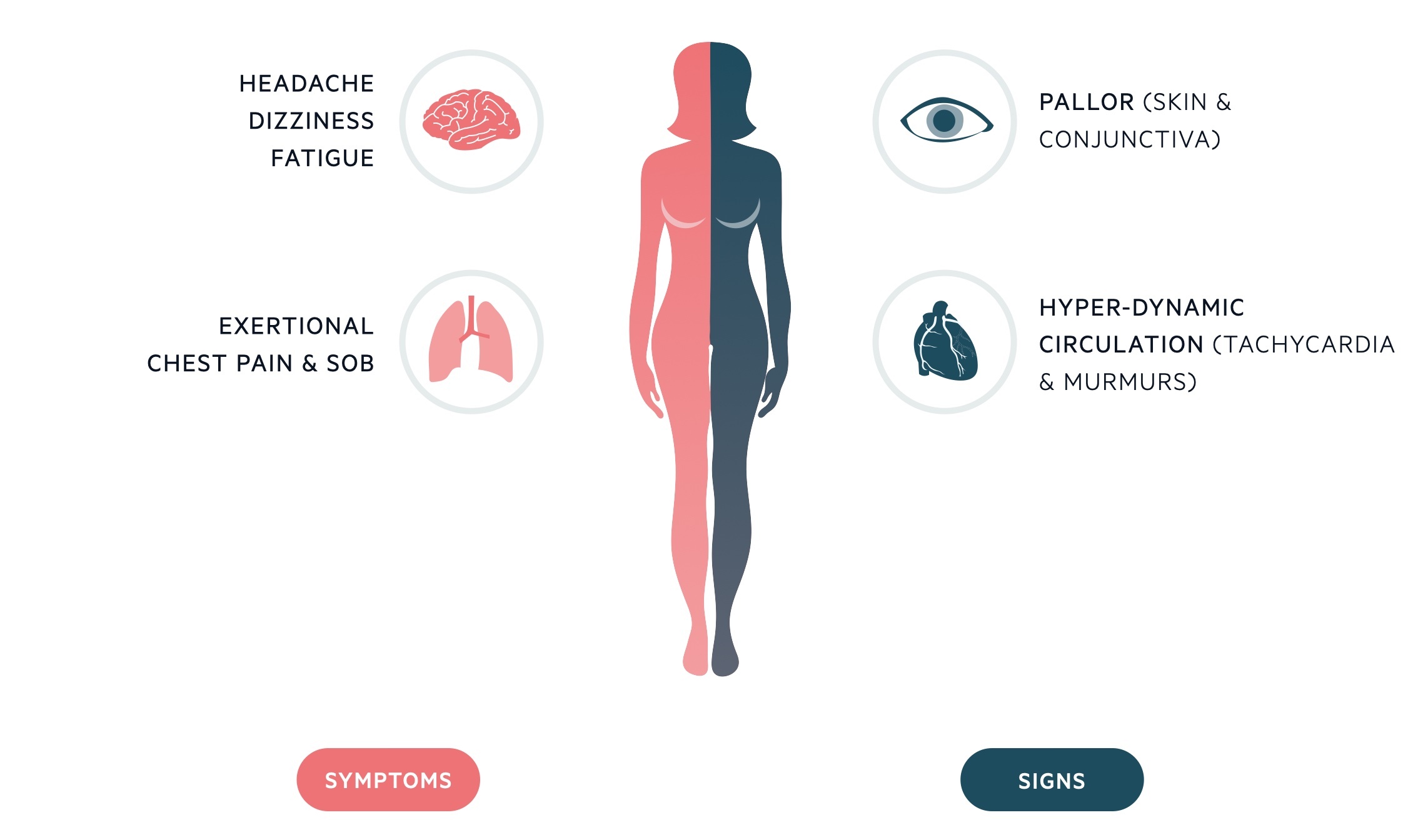
Due to the chronicity of IDA, symptoms generally occur at low levels of haemoglobin concentration (e.g. < 80 g/L). These include fatigue, dyspnoea, dizziness, headache, nausea, bowel disturbance, and possible exacerbation of cardiovascular co-morbidities causing angina, palpitations, and intermittent claudication.
Classical signs of IDA include glossitis, koilonychia (spoon-shaped nails), angular stomatitis, and conjunctival pallor.
Interestingly, some children with IDA may be found to have peculiar dietary cravings (pica) for materials such as soil, clay, or chalk. Whether this is a cause or consequence of IDA is not fully understood.
Investigations
FBC, haematinics, blood film and iron studies help diagnose and evaluate severity.
Iron studies include: serum iron, ferritin, and iron-binding globulin (transferrin).
The FBC is useful in the diagnosis of microcytic anaemia (e.g. low Hb & low MCV) but further investigations are needed to determine the underlying cause.
The FBC also provides the mean corpuscular haemoglobin concentration (MCHC). A low MCHC is reflected in the blood film as small, pale erythrocytes that may show some alteration in shape (e.g. pencil-shaped cells).
Serum iron, ferritin, and transferrin levels are essential in the diagnosis of IDA.
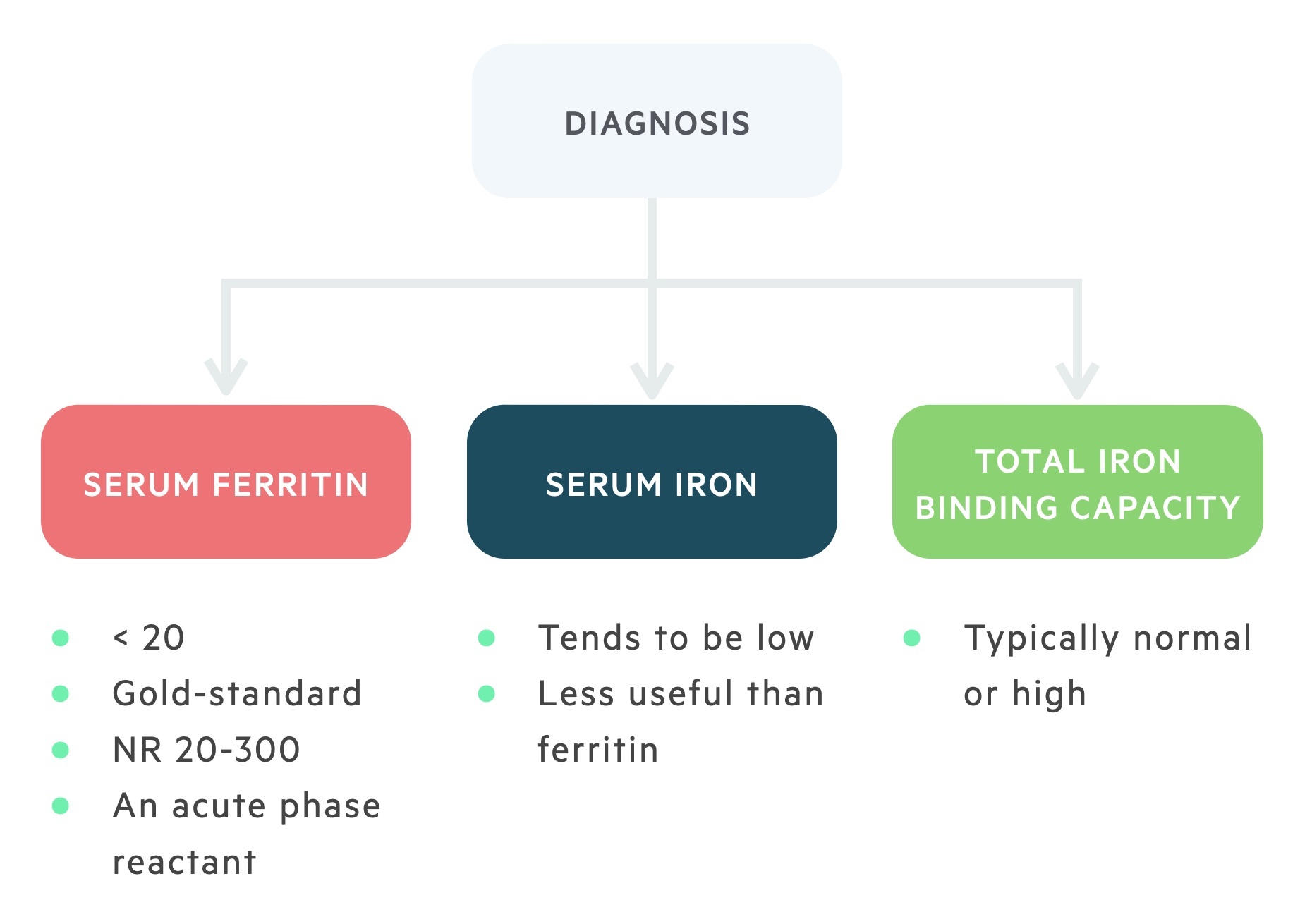
Serum iron and ferritin are usually low suggesting a depletion of iron stores. On the other hand, transferrin levels are normal or increased reflecting an increase in total iron-binding capacity (i.e. there is a lack of iron-binding to transferrin increasing its binding potential). Due to the decrease in iron-binding to transferrin, there is a reduction in transferrin saturation.
Once iron-deficiency is confirmed, further investigations are directed at trying to establish the cause. These include:
- Coeliac screening (immunoglobulins and anti-tissue transglutaminase)
- Urinalysis (assess for urinary tract pathology)
- Upper & lower GI endoscopy (in appropriate cases - see below)
- Small bowel capsule endoscopy (considered if negative bidirectional endoscopy and inadequate response to iron or recurrence)
Management
Management of IDA should involve investigation into the underlying cause & replacement of iron.
Male and postmenopausal female patients with unexplained IDA should be investigated for suspected gastrointestinal malignancy (or indeed benign GI pathology). The upper gastrointestinal tract is more commonly the site of pathology than the lower gastrointestinal tract.
Oral replacement of iron in the form of ferrous fumarate or ferrous sulfate are common pharmacotherapies. Follow-up blood tests should always be completed to assess for response to treatment and patients should be warned about side-effects. These may include constipation, black stools, diarrhoea, nausea, and dyspepsia/epigastric discomfort. For those who are unable to tolerate oral iron an infusion of iron can be arranged. Treatment should continue for 3 months to ensure adequate replacement of stores.
Iron replacement should generally be offered as once a day. This can be reduced to alternative days if there are side-effects. Importantly, if a limited red blood cell transfusion is needed for severe, symptomatic anaemia then iron replacement is still necessary afterwards.
Anaemia of chronic disease
Anaemia of chronic disease (ACD) is a complex and multi-factorial condition due to a chronic inflammatory process from underlying infection, malignancy or systemic disease.
ACD is the second most common cause of anaemia worldwide, and commonly seen among hospitalised patients. ACD is essentially a functional iron deficiency (FID) whereby the supply of iron for erythropoiesis (i.e. generation of new red blood cells) is inadequate despite apparently normal cellular iron stores.
Aetiology & pathophysiology
ACD is classically described as a normocytic, normochromic anaemia secondary to systemic diseases, infection or malignancy.
The pathophysiology of ACD is complex and a number of mechanisms have been proposed including altered hepcidin regulation, inhibition of erythropoiesis, low erythropoietin levels and increased phagocytosis of erythroid cells. ACD or FID is commonly seen in patients with chronic kidney disease (CKD) and chronic heart failure (CHF).
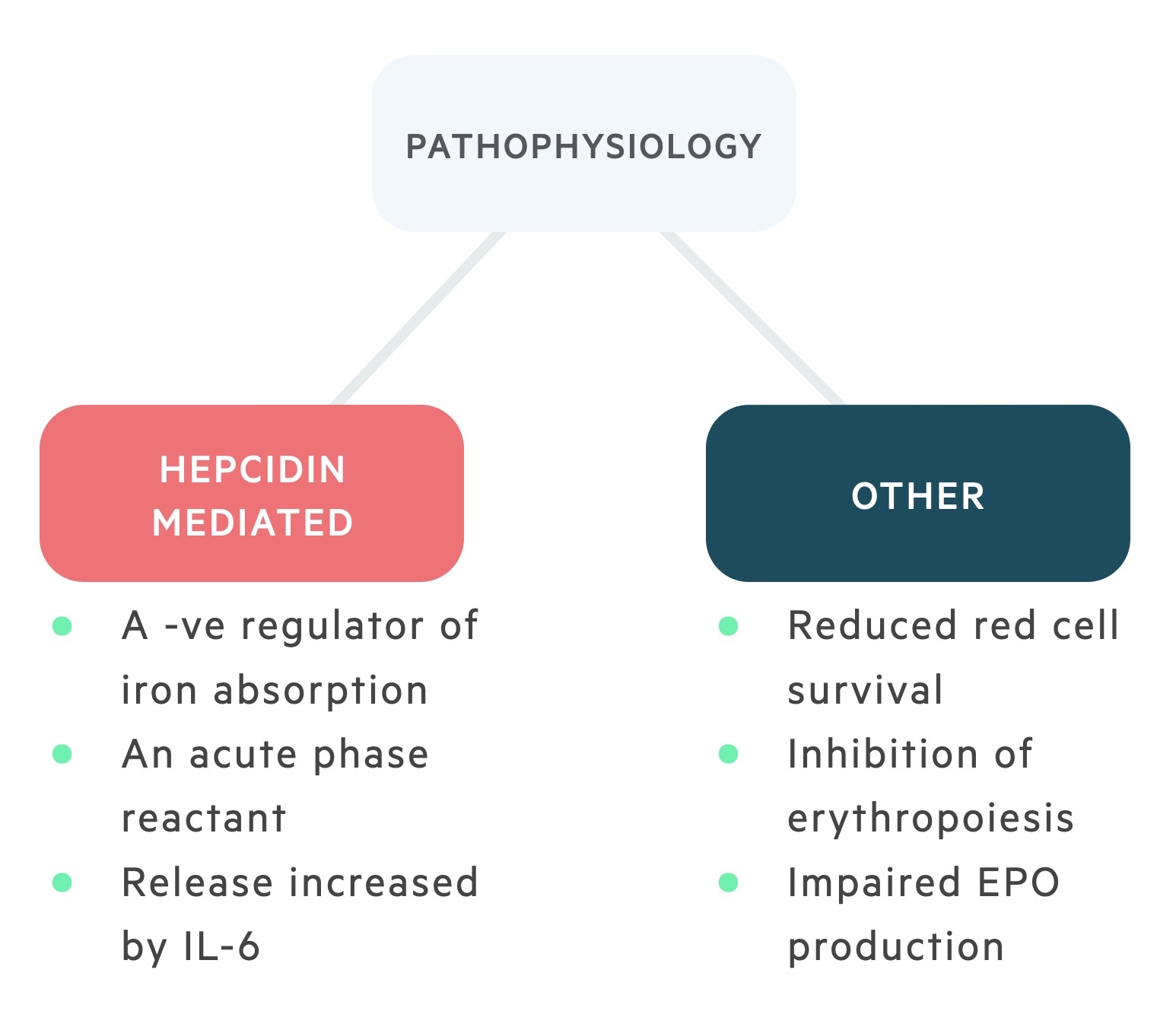
Hepcidin is the normal regulator of iron absorption from enterocytes and the tissue distribution of iron. It is an acute phase protein that usually works to reduce the availability of iron from infecting microorganisms.
Chronic inflammation mediated by IL-6 can lead to a hepcidin-induced block of iron absorption and iron release from macrophages. This reduction in the availability of iron for the production of erythrocytes as part of erythropoiesis can lead to a microcytic anaemia. However, this is only seen in 25% of cases.
Clinical features
The clinical presentation of ACD is generally that of the underlying disorder, and unless the haemoglobin concentration is severely reduced, patients may be asymptomatic.
If present, clinical features are typical of all anaemias including fatigue, headache and dizziness. If severely symptomatic, patients may develop palpitations, angina or dyspnoea.
Investigations
Key investigations in ACD include the FBC, CRP, haematinics, iron studies and blood film.
The FBC may show a normocytic normochromic anaemia (approx. 75%) or the development of a microcytic anaemia (approx. 25%). In ACD, the MCV is rarely below 70 fL.
Similar to IDA, serum iron, ferritin and transferrin are vital for the diagnosis of ACD.
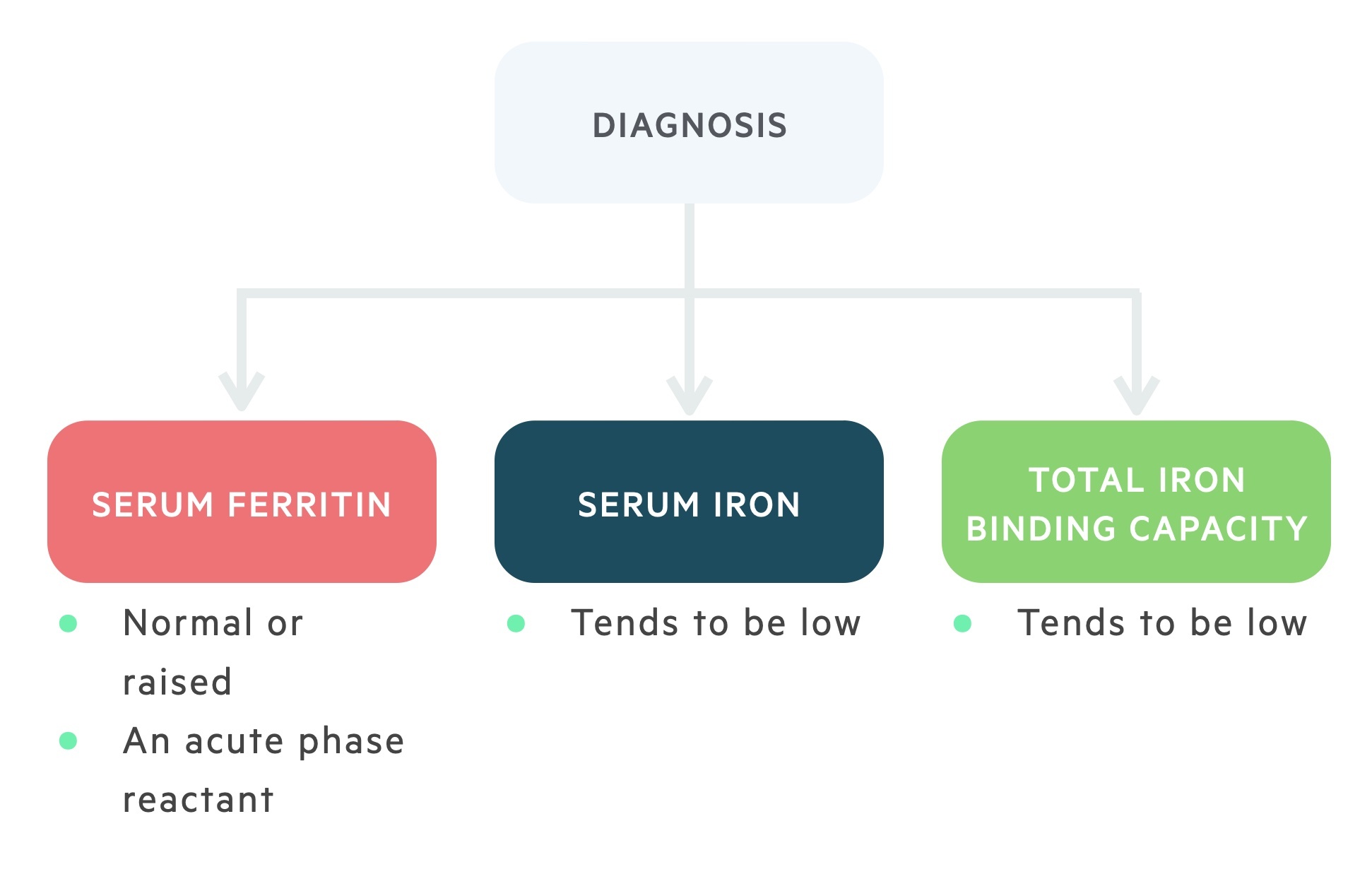
Ferritin, like hepcidin, is an acute phase reactant meaning its levels typically increase in chronic inflammation. Therefore, in ACD ferritin levels may be normal or increased.
Transferrin and serum iron levels tend to be reduced. The normal or decreased level of transferrin is a differentiating factor from IDA.
Another differentiating factor is marrow iron stores (evaluated with a biopsy). In IDA, iron stores are absent, whereas in ACD they are normal or increased.
Management
As mentioned, ACD is a multifactorial disorder that has a number of management options depending on the degree of anaemia, severity of symptoms and the underlying diagnosis.
In general, management should involve treatment of the underlying disorder and correct management of any complicating factors including iron, B12 and folate deficiency.
Other options for the treatment of ACD can include the use of erythropoietin, parenteral iron and transfusions as indicated. ACD or FID is important to recognise in patients because it responds poorly to oral iron replacement therapy.
Beta-thalassaemia
The beta-thalassaemias are a heterogenous group of conditions that result from a reduced or absent production of beta-globin chains.
The prevalence of the beta-thalassaemias varies widely depending on the ethnic population. Beta-thalassaemia is seen more commonly in African, Mediterranean and South-east Asian populations.
Aetiology & pathophysiology
Normal adult haemoglobin (HbA) is composed of two alpha-globin chains and two beta-globin chains.
Beta-thalassaemias occur due to genetic mutations within the beta-globin genes, which are located on the short arm of chromosome 11.
This gene is termed the haemoglobin subunit beta (HBB) and it is essential for normal beta-globin chain production.
There are many hundreds of gene mutations within the HBB that lead to the development of thalassaemia. In general, mutations can lead to an absence of production (beta 0 thalassaemia) or a reduced production (beta + thalassaemia) of the beta-globin chain.
Patients with two abnormal alleles (homozygous) are said to suffer from beta-thalassaemia major and will have an absent or severely reduced production of beta-globin chains.
Patients with one abnormal allele (heterozygous) are said to suffer from beta-thalassaemia minor or ‘trait’ and have a mild reduction in beta-globin chain production.
Reduced/absent production leads to an excess in alpha-globin chains. These excess alpha-globin chains precipitate within the cells as they are unstable and incapable of forming normal tetramers. The degree of alpha-globin chain excess determines the severity of the clinical presentation.
Clinical features
Patients with beta-thalassaemia major tend to present in the first year of life as the production of foetal haemoglobin (HbF: two alpha / two gamma chains) is replaced by defective adult haemoglobin.
Clinical features tend to reflect the severity of beta-thalassaemia. Patients with beta-thalassaemia major tend to have marked extramedullary haematopoiesis and complications secondary to iron overload from repeated transfusions.
- Extramedullary haematopoiesis
- Hepatomegaly
- Splenomegaly
- Skeletal abnormalities
- Iron overload
- Hypogonadism
- Growth failure
- Diabetes mellitus
- Hypothyroidism
Patients with beta-thalassaemia minor/trait are usually entirely asymptomatic and found to have a microcytic anaemia on routine testing.
Investigation
The diagnosis of beta-thalassaemia is confirmed with haemoglobin electrophoresis.
Hb electrophoresis shows reduced or absent levels of HbA and the presence of increased HbF and HbA2. The latter is composed of two alpha chains and two delta chains. Importantly, HbA may be a present in patients who have recently been transfused.
Other laboratory tests are important in the workup of the beta-thalassaemias and involve an FBC, blood film, iron studies, haematinics, LDH, bilirubin (as part of LFTs) and haptoglobin.
The predominant finding is a profound microcytic anaemia (MCV < 75 fL) with evidence of microcytic, hypochromic erythrocytes on blood film and normal iron studies.
Management
Management is variable and depends on the severity of beta-thalassaemia.
In general, patients with beta-thalassaemia trait do not require any treatment.
Patients with beta-thalassaemia major will require early, frequent blood transfusions. Other management options include splenectomy and hematopoietic stem cell transplantation. Patients receiving recurrent transfusions may suffer with iron overload and require iron chelation therapy.
Uncommon causes
Other causes of microcytic anaemia include sideroblastic anaemia, lead poisoning and other haemoglobinopathies.
Sideroblastic anaemia can be congenital (X-linked recessive) or acquired. It results in the inclusion of a perinuclear ring of coarse iron-containing granules (‘ring-sideroblasts’) within erythrocyte progenitors.
Acquired causes may be seen in myelodysplastic syndromes or following the use of pharmaceuticals such as isoniazid or chloramphenicol.
Lead poisoning is an uncommon cause of microcytic anaemia that can cause a reduction in the synthesis of haem and a reduced lifespan of erythrocytes.
Last updated: January 2023
Have comments about these notes? Leave us feedback