Shock
Notes
Introduction
Shock describes circulatory failure resulting in inadequate tissue perfusion and insufficient delivery of oxygen.
Shock is a broad and at times unhelpful term. It refers to any cause of circulatory failure that results in inadequate oxygen delivery to tissues. There are many causes with different underlying pathophysiological processes, normally they are divided into four categories:
- Hypovolaemic
- Distributive
- Cardiogenic
- Obstructive
It should be noted the term ‘shock’ is unliked by many in the medical field. The Royal College of Emergency Medicine describe it as a ‘somewhat lazy shorthand’ preferring clinicians take a more granular approach to their description of the patients clinical state that better reflects the causes and mechanisms. The term remains widely used.
Basic sciences
Oxygen delivery is determined by two major factors; cardiac output and arterial oxygen content.
Cardiac output
Cardiac output is determined by the heart rate and stroke volume. Stroke volume refers to the amount of blood pumped out of the heart from each contraction. Cardiac output refers to the amount of blood pumped out of the heart in one minute, equivalent to HR x SV.
The heart is responsible for maintaining adequate cardiac output through rate and stroke volume. With an average of 70 bpm and 70 ml per beat, cardiac output can be estimated at 5L / min.

Heart rate is predominantly determined by the sympathetic nervous system and parasympathetic nervous system (autonomic nervous system).
Stroke volume is determined by a complex interplay of preload, myocardial contractility and afterload:
- Preload - stretching of cardiomyocytes at the end of diastole.
- Myocardial contractility - changes to stroke volume can be brought about through changes to contractility.
- Afterload - pressure or load against which the ventricles must contract.
Oxygen delivery
Circulation allows for the delivery of oxygen to tissues (and removal of waste products) with the ability to make adjustments depending fluctuating oxygen consumption.
Oxygen is carried in the blood in two ways:
- Bound to haemoglobin
- Dissolved in plasma (small amount)
Haemoglobin carries the vast majority of oxygen in the blood. It consists of four globin chains (normally two alpha and two beta chains) each with one haem group containing ferrous iron. Each haemoglobin can bind to four oxygen molecules.
An estimate of oxygen delivery to tissues can be obtained with the following equation:
(HR x SV) x [Hb]g/dl x 10 x 1.34 x sO2 ml/l
See RCEM website for more details.
Clinical features and compensation
The body has a number of compensatory techniques to deal with a fall in cardiac output or inadequate tissue perfusion. The compensatory mechanisms that are activated are in a large part down to the underlying cause of the shock, though many are common to most causes.
Some of the systems that may become activated include:
- Baroreceptors: found at the carotid sinus and aortic arch, they respond to falls in blood pressure by increasing sympathetic output and reducing vagal tone. The increased sympathetic output causes heart rate to increase, vasoconstriction and stimulation of adrenaline and noradrenaline release from the adrenal medulla.
- Renin-angiotensin-aldosterone system: a reduction in renal perfusion that accompanies a fall in blood pressure activates the RAA system. The release of angiotensin II causes vasoconstriction and aldosterone causes reabsorption of fluid by the kidneys.
- Chemoreceptors: poor tissue perfusion results in increased lactic acid production secondary to anaerobic respiration. A reduction in the pH of blood activates chemoreceptors to trigger an increase in respiratory rate. The increased respiratory rate leads to more carbon dioxide being ‘blown off’ - this is the basis for respiratory compensation in response to metabolic acidosis.
The clinical features of shock are determined by the underlying cause. In hypovolaemic shock patients will typically be tachycardic and peripherally ‘shut down’. This refers to cool peripheries and a prolonged capillary refill secondary to vasoconstriction. Both the increased heart rate and peripheral vasoconstriction are compensatory responses to reduced blood volume. Conversely in distributive shock peripheral vasodilation is the common aetiological factor. These patients may be warm peripherally though this may change, particularly in later stages. Tachycardia is again commonly seen.
Respiratory rate is a sensitive sign of systemic upset, tachypnea often occurs as a compensatory mechanism in patients with a metabolic acidosis secondary to inadequate tissue perfusion. Urine output is essential to monitor - oliguria is common - reduced urine output reflecting the compensatory and fluid preserving actions of aldosterone and vasopressin. Mental status should be recorded and monitored, as shock worsens confusion, reduced responsiveness and coma may result.
Decompensation
Decompensation occurs when normal compensatory mechanisms are overwhelmed and can no longer allow the body to maintain adequate blood pressure and tissue perfusion. This leads to a complex cascade of life-threatening failures.
Increasing anaerobic respiration results in greater acid production at a time when reduced renal perfusion and function prevents excretion of hydrogen ions. Acidosis has a negative impact on many of the bodies functions including normal cardiac function.
In severe cases, fall in blood pressure results in vascular stasis in small vessels and the release of local inflammatory mediators. Widespread clotting leads to a consumptive coagulopathy (i.e. coagulopathy secondary to clotting factors being used up) causing disseminated intravascular coagulation (DIC).
End-organs begin to fail in response to inadequate oxygen supply. Ischaemic liver injury results in the release of hepatic enzymes, causing startling rises in ALT and AST. Acute tubular necrosis results in renal failure worsening acidosis and electrolyte abnormalities (e.g. hyperkalaemia).
Hypovolaemic shock
Hypovolaemic shock occurs secondary to a reduction in intravascular volume.
The causes of hypovolaemic shock can be divided into haemorrhagic and non-haemorrhagic. The features of such volume loss depend on the rate of loss, the patients co-morbidities and their ability to compensate. Reduced intravascular volume results in low blood pressure and poor tissue perfusion:
- Low blood pressure: stimulates baroreceptors to increase sympathetic output, osmoreceptors in the hypothalamus to release ADH and the kidneys to activate the RAA system. The result of increased sympathetic output is vasoconstriction, tachycardia and noradrenaline/adrenaline release (further accentuating the sympathetic response). ADH release results in greater fluid retention through reduced excretion in the kidneys. Activation of RAA leads to the release of angiotensin II which prompts vasoconstriction and aldosterone which causes reabsorption of fluid in the kidneys.
- Poor tissue perfusion: this leads lack of oxygen delivery and anaerobic respiration with lactic acid production. These factors drive chemoreceptors to increase the respiratory rate to allow the ‘blowing off’ of carbon dioxide - to compensate (or attempt to) for the metabolic acidosis.
Patients are normally tachycardic and peripherally ‘shut down’. This refers to cool peripheries and a prolonged capillary refill secondary to vasoconstriction. Both the increased heart rate and peripheral vasoconstriction are compensatory responses to reduced blood volume.
Haemorrhagic
Haemorrhagic shock results from volume loss due to acute blood loss. Causes include trauma, self harm, acute GI bleed, obstetric haemorrhage, aneurysm rupture and post-operative bleeding.
The severity of haemorrhagic shock can be classified based on a number of parameters.
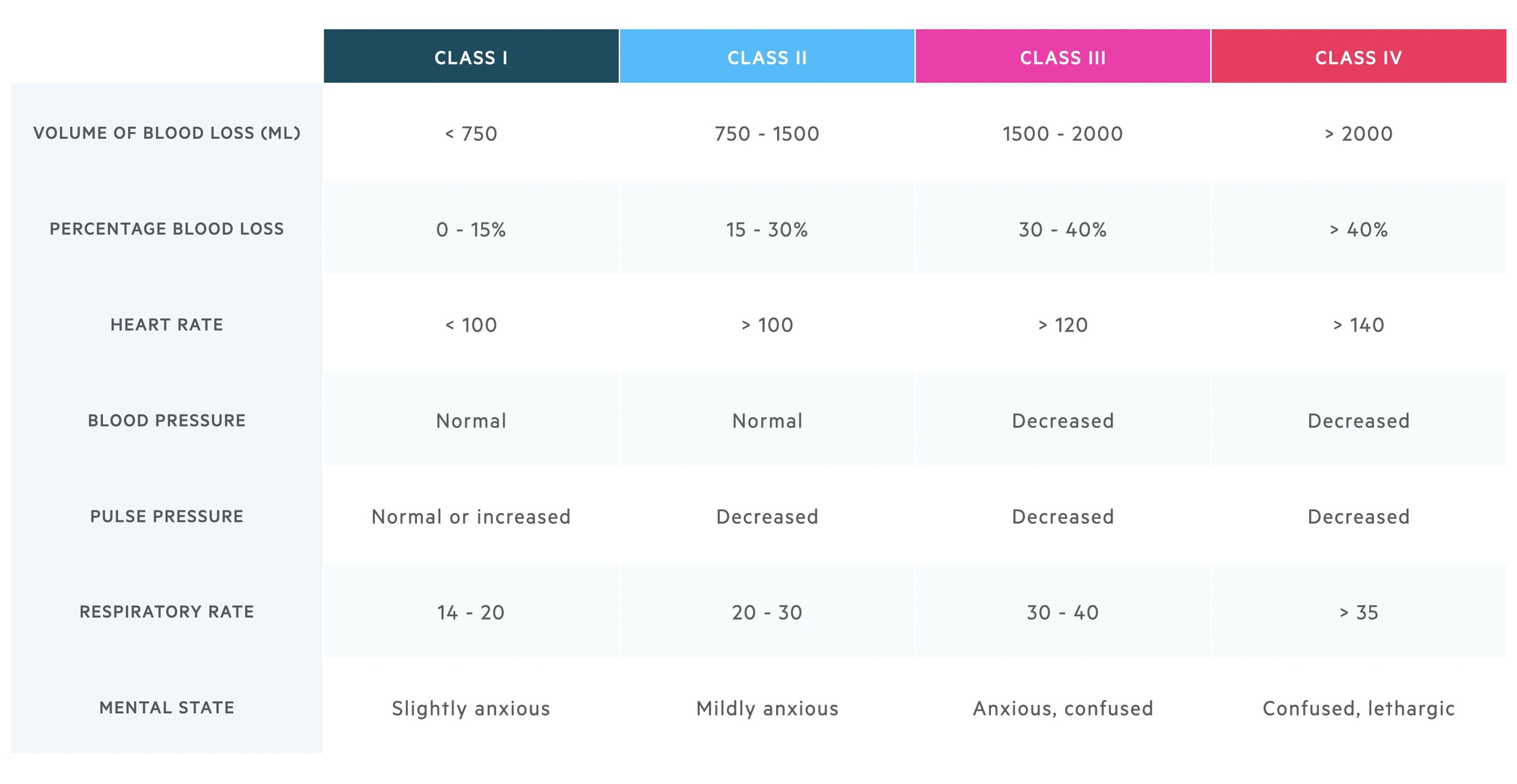
This table reveals a number of interesting pieces of information. Blood pressure is not a particularly sensitive sign of acute volume loss, whilst respiratory rate increases relatively early on. Remember these findings give a general outline but individual patients may deviate.
Non-haemorrhagic
Non-haemorrhagic shock occurs due to reduced volume from increased fluid losses or reduced intake. Examples include DKA, severe burns and severe diarrhoea and vomiting.
Distributive shock
Distributive shock is caused by peripheral vasodilatation leading to abnormal volume distribution and inadequate perfusion.
Distributive shock has many causes, some texts consider three distinct categories (vasodilatory, anaphylactic and neurogenic) in the place of distributive. Here we discuss three important examples.
Septic shock
Septic shock describes shock secondary to sepsis. Sepsis can be thought of as a dysregulated host response to infection leading to life-threatening organ dysfunction. Features include haemodynamic instability, tachypnea, changes to mental status, reduced urine ouput and pyrexia.
There are a number of scoring systems that can be used to assess risk and prognosis, each has their own limitations. An example is qSOFA (quick Sepsis Related Failure Assessment) that aims to identify patients at increased risk of poor outcomes outside the ITU environment. It consists of three components:
- Mental status (score 1 if altered mental status)
- Respiratory rate (score 1 if ≥ 22)
- Systolic BP (score 1 if ≤ 100)
Any patient with suspected sepsis should be managed with the SEPSIS-6 principles in mind. The sepsis six bundle is a protocol by which a patient may be investigated and treated for sepsis. It is by no means exhaustive but incorporates key investigation and treatment points. The bundle should be initiated and completed within one hour of recognition of the signs. All patients with significant infection should have a senior review. Further support may be required including vasopressors (e.g. noradrenaline) to maintain blood pressure, given in AE resus or ITU.

Three In
Patients should receive high flow oxygen to achieve appropriate target saturations. A target of 94-98% is appropriate for the majority of patients. Those at risk of carbon dioxide retention (COPD) should have a target of 88-92%. IV fluids should be started, often 500ml of crystalloid over 15 minutes with reference the patient's haemodynamic status and co-morbidities. Antibiotics are key and should be given without delay. The choice should be in line with local hospitals guidelines. There are usually recommendations for treating 'sepsis of unknown cause' if the site of infection is unknown.
Three out
A minimum of two sets of blood cultures should be taken. Ideally, this should happen prior to administration of antibiotics though they should not be a cause for delay. A serum lactate should be obtained normally via a blood gas (arterial or venous) to help assess the patient's status. Urine output should be measured ideally with a catheter with a careful fluid balance recorded.
Neurogenic shock
Brain and spinal cord injuries can cause failure of normal autonomic pathways. If caused by a spinal cord injury these tend to be above T6. A failure of sympathetic tone and unopposed vagal tone results in vasodilation, reduced venous return and a fall in cardiac output. Bradycardia may also play a role.
Anaphylactic shock
Describes a severe allergic reaction, caused by a type I hypersensitivity reaction mediated by IgE release. It results in hypotension, laryngeal oedema and bronchoconstriction.
For more see our Anaphylaxis notes.
Cardiogenic shock
Cardiogenic shock is caused by cardiac dysfunction that leads to inadequate cardiac output.
Cardiogenic shock can result from any cardiac cause of reduced output. Arrhythmias, valvular pathologies (e.g. acute aortic regurgitation) and myocardial infarction can all lead to cardiogenic shock.
Obstructive shock
Obstructive shock is caused by mechanical obstruction causing a failure of adequate cardiac output.
There are a number of causes of obstructive shock which include:
- Pulmonary embolism: large PEs (e.g. saddle embolus) can cause right sided heart failure due to elevated pulmonary vascular resistance.
- Tension pneumothorax: elevated pressure in the pleural cavity compresses the heart and great vessels leading to obstruction of venous return and ventricular filling. This results in a failure of cardiac output.
- Cardiac tamponade: this has a number of effects on normal physiology but can be simply thought of as restricting venous return and ventricular filling.
Last updated: July 2021
Have comments about these notes? Leave us feedback