Multiple myeloma
Notes
Overview
Multiple myeloma refers to a malignant disorder of plasma cells (mature B lymphocytes).
Multiple myeloma (MM) is the second most common haematological malignancy. It is characterised by excess secretion of a monoclonal antibody. We term it a monoclonal antibody, because it is derived from a single clone of plasma cells that have undergone abnormal proliferation.
MM accounts for 60-70 cases per 1,000,000 people each year, although the overall prevalence of the condition is increasing due to the improved survival with newer treatments. Unfortunately, it still accounts for 2% of cancer-related deaths and it is associated with a number of severe complications including spinal cord compression, renal impairment and hypercalcaemia,
Immune response
Our immune system is broadly divided into the innate and adaptive immune response.
All blood cells are derived from haematopoietic stem cells (HSCs), which differentiate into three main cell lineages: erythroid, myeloid, lymphoid. See our notes on haematopoiesis.
We have a variety of different cell types that are important in both innate (e.g. neutrophils) and adaptive (e.g. lymphocytes) immunity. The chemical messengers of the immune system are small molecules, such as cytokines, which are released by a variety of cell types including immune cells and endothelial cells.
Innate immunity
This is our first line of defence against microorganisms, which involves both cells (e.g. phagocytes, dendritic cells) and molecules (e.g. complement, cytokines). It involves recognition of foreign material by identification of conserved constitutes of microorganisms. We call these pathogen-associated molecular patterns (PAMPs). This stimulates a pro-inflammatory response and activates the adaptive immune response.
Adaptive immunity
This immune response is divided into cellular immunity and antibody-mediated immunity. There are three hallmarks of adaptive immunity:
- Antigen specificity: it can detect almost an unlimited number of antigens with high accuracy.
- Delayed onset: takes 2-4 days to mount an adaptive immune response (on first encounter).
- Immune memory: rapidly protective on re-exposure to a foreign antigen.
Lymphocytes are the primary immune cell in adaptive immunity. T lymphocytes are important in both cellular immunity and mediating the antibody response (through activation of B lymphocytes). B lymphocytes are important in antibody-mediated immunity, which mature into plasma cells.
Antibody-mediated immunity
The secretion of antibodies forms a key part of our adaptive immune response.
B lymphocytes are the antibody secreting cells in the body. Immature cells mature within the bone marrow. On exposure to an antigen, naive B cells mature into plasma cells.
Antibodies
Antibodies are composed of heavy and light chains. There are five types of heavy chain (A, G, M, D, E) and two types of light chain (Kappa, lambda). Two heavy chains form with two light chains to create the complete antibody.
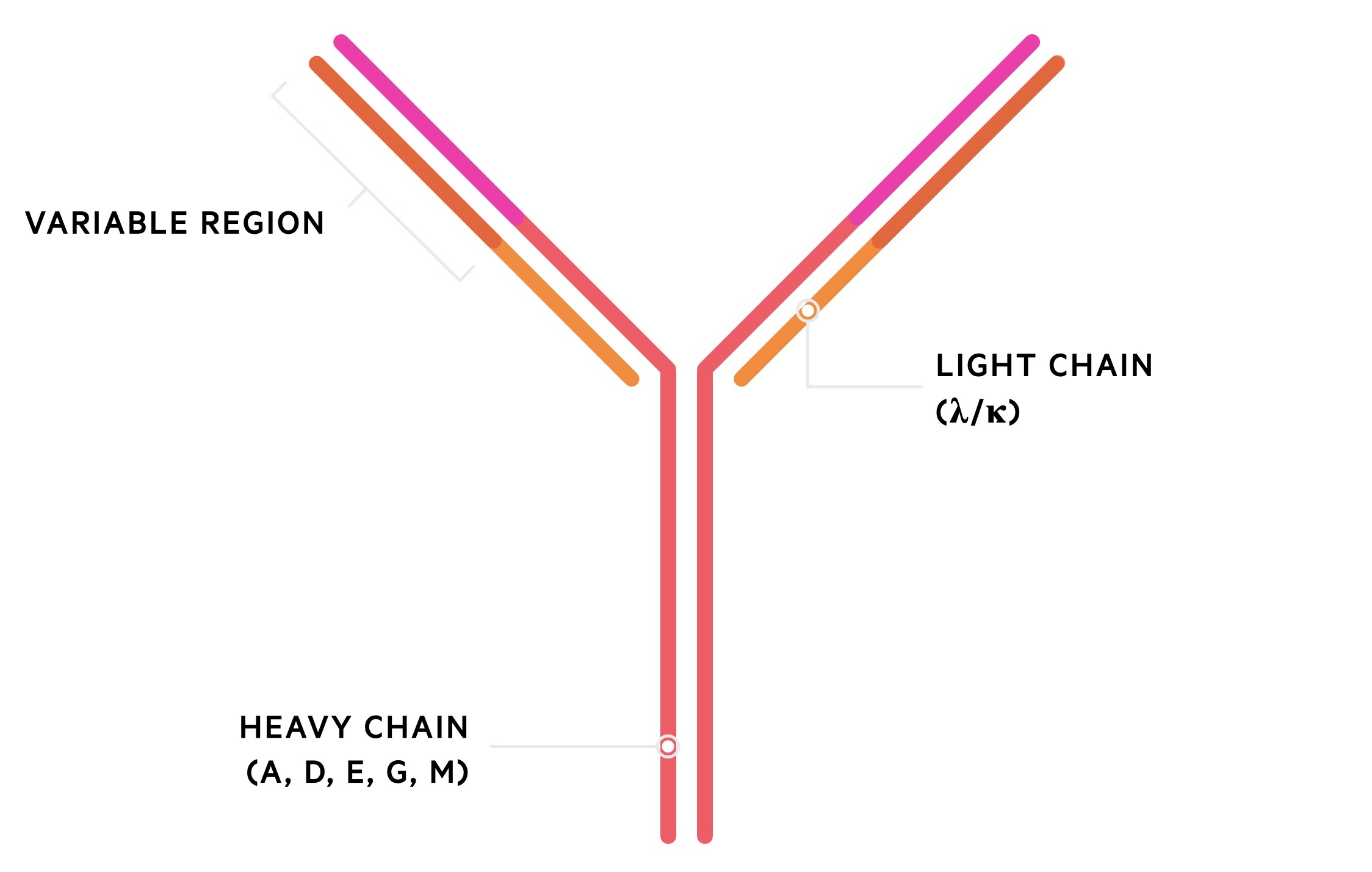
Maturation and proliferation
The body contains thousands of antigen-specific B cells. Once activated they multiply into a clone of plasma cells that secrete a specific antibody to a foreign antigen. Collectively, the pool of antibodies from all the different clones of B cells are termed polyclonal antibodies (i.e. each antibody has a different antigen-specificity).
If one clone acquires an abnormal mutation and proliferates uncontrollably, it will secrete its own antigen-specific antibody in excess of the others. These are termed monoclonal antibodies (i.e. a group of the same antibodies from a clone plasma cells). These monoclonal antibodies are associated with a single light chain (kappa/lambda).
Aetiology and pathophysiology
The pathophysiology of MM is still poorly understood but there appears to be a two-step model.
The pathophysiology of MM is centred around the development of a malignant clone of plasma cells, which can secrete excess amounts of a monoclonal antibody. This is due to the development of cytogenetic abnormalities, which refers to structural chromosomal changes, mutations or cellular dysregulation (e.g. cell cycle or apoptosis dysregulation).
There appears to be two key steps in the pathophysiology of MM: development of monoclonal gammopathy of undetermined significance (MGUS) and progression from MGUS to MM.
- Development of MGUS: almost all cases of MM arise from this premalignant plasma cell disorder. Affects 3% of patients > 50 years old. Initial cytogenetic abnormality occurs (inciting event). Thought to be an abnormal plasma cell response to antigen stimulus. Leads to creation of plasma cell clone that secretes a monoclonal antibody. Most will not develop MM.
- Progression from MGUS to MM: rate of progression estimated at 1% per year. Further cytogenetic abnormalities and changes to the bone marrow microenvironment occur, which promotes proliferation. Associated with systemic problems due to plasma cell infiltration of bone marrow and excess light chain secretion.
There is a further intermediate stage between MGUS and MM. This known as smouldering myeloma or asymptomatic myeloma. It is a more advanced premalignant stage due to a higher burden of clonal plasma cells in the bone marrow.
Clinical presentations
The clinical presentation of MM is generally related to the infiltration of plasma cells and secretion of monoclonal antibodies.
Patients with MM may have constitutional features of malignancy including weight loss, fatigue, loss of appetite and/or generalised weakness. There are a number of typical clinical presentations that relate to the excess proliferation and infiltration of plasma cells (usually in bone marrow) and the excess secretion of monoclonal antibodies.
Presenting clinical features
- Bone disease: widespread due to clonal proliferation in bone marrow. Seen as lytic lesions on imaging. Can lead to fractures.
- Impaired renal function: >50% have raised creatinine at diagnosis. Kidneys affected in multiple ways. Commonly due to light chain nephropathy (tubules blocked by light chain casts).
- Anaemia: seen in >90% at some point during disease course. Normal bone marrow destroyed by proliferation of malignant plasma cells. Renal disease may contribute (EPO deficiency).
- Hypercalcaemia: MM-induced bone demineralisation. More common in active disease. At high levels (≥ 2.9 mmol/L) should be treated as a medical emergency. See our notes on hypercalcaemia.
- Recurrent or persistent bacterial infection: immune dysfunction and hypogammaglobulinaemia due to suppression of normal plasma cell function.
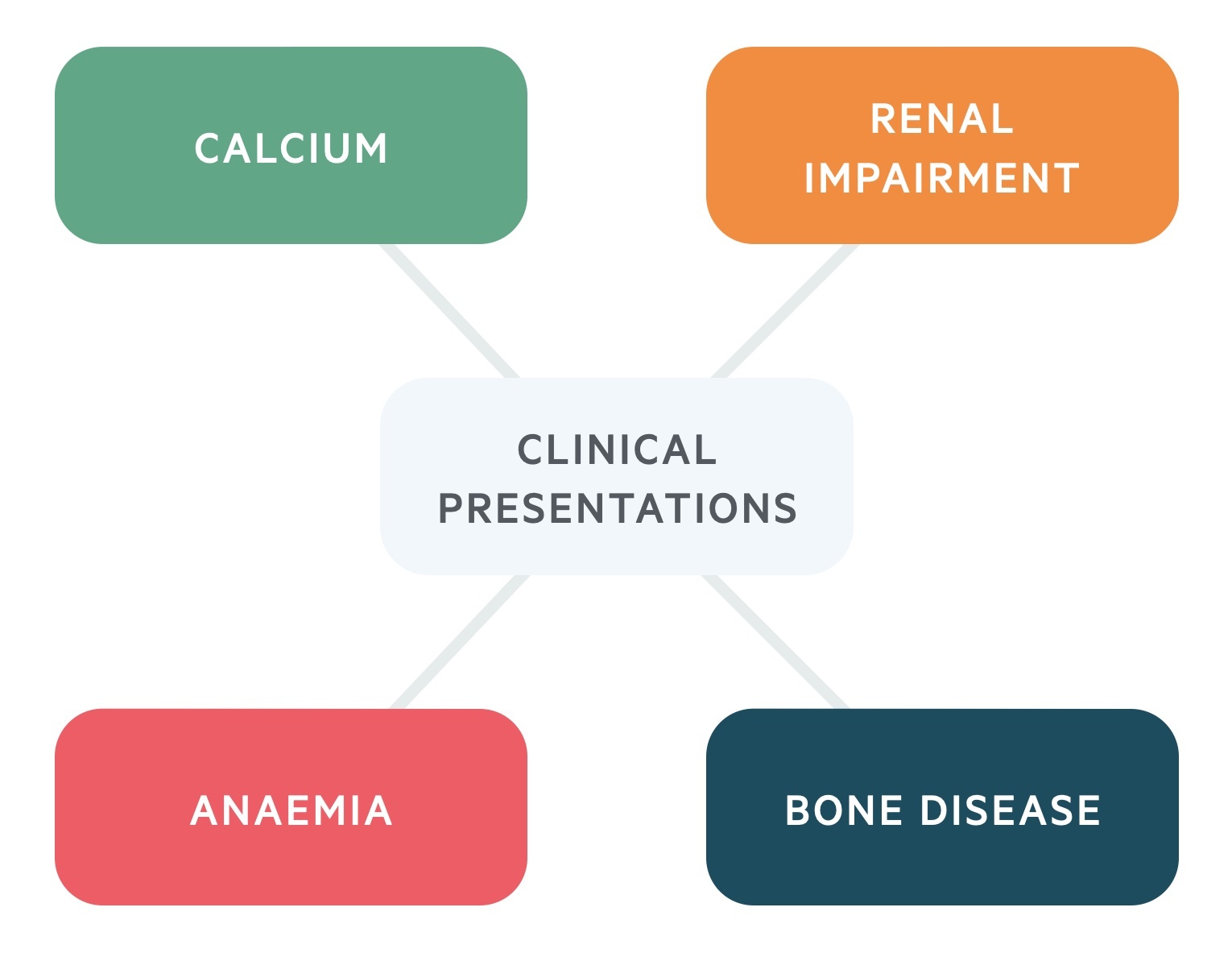
Mnemonic
These key clinical features of MM can be remembered using the mnemonic ‘CRAB’.
- C - calcium levels high
- R - renal impairment
- A - anaemia
- B - bone disease
Other syndromes
MM can present with a myriad of other clinical features, some presenting as medical emergencies. These may include paraesthesia, fever (<1%), splenomegaly (1%), hepatomegaly (4%) or lymphadenopathy (1%). Neurological involvement can result from hyperviscosity syndromes, spinal cord compression, peripheral neuropathy or radiculopathy.
- Hyperviscosity syndrome: may develop with high paraprotein levels (i.e. high IgA or IgG). Typical symptoms include blurred vision, headaches, mucosal bleeding and dyspnoea due to heart failure. Requires urgent plasma exchange.
- Spinal cord compression: can occur in 5% of patients during course of disease. Highly variable depending on lesion causing compression, location, and rate of development. See our notes on cord compression.
When to suspect myeloma
There should be a low threshold for myeloma screening in any patient with suggestive clinical features.
Myeloma should be suspected in any patient with typical features, but particularly those over 60 years old with:
- Unexplained bone pain (and pathological fractures)
- Fatigue
- Symptoms of hypercalcaemia: bone pain, abdo pain, constipation, confusion, polyuria
- Weight loss
- Symptoms of cord compression: back pain, new leg weakness, bladder/bowel dysfunction
- Symptoms of hyperviscosity: headache, blurred vision, shortness of breath, mucosal bleeding
- Recurrent infections
Screening for myeloma
‘Screening’ for myeloma involves looking for monoclonal antibodies, which are the secretion product of the malignant clones.
When we ‘screen’ for myeloma we are looking for the secretion product of the malignant clone of plasma cells - the monoclonal antibodies. We can do this using protein electrophoresis and immunofixation. Electrophoresis tells us whether there is an increased number of antibodies. This is followed by immunofixation, which tells us what type of antibody has increased (i.e. is it a monoclonal antibody). MM is usually the result of IgG, IgA or the accompanying light chain. It rarely occurs with IgM.
NOTE: The presence of an IgM monoclonal antibody suggests another haematological malignancy termed Waldenstrom macroglobulinemia.
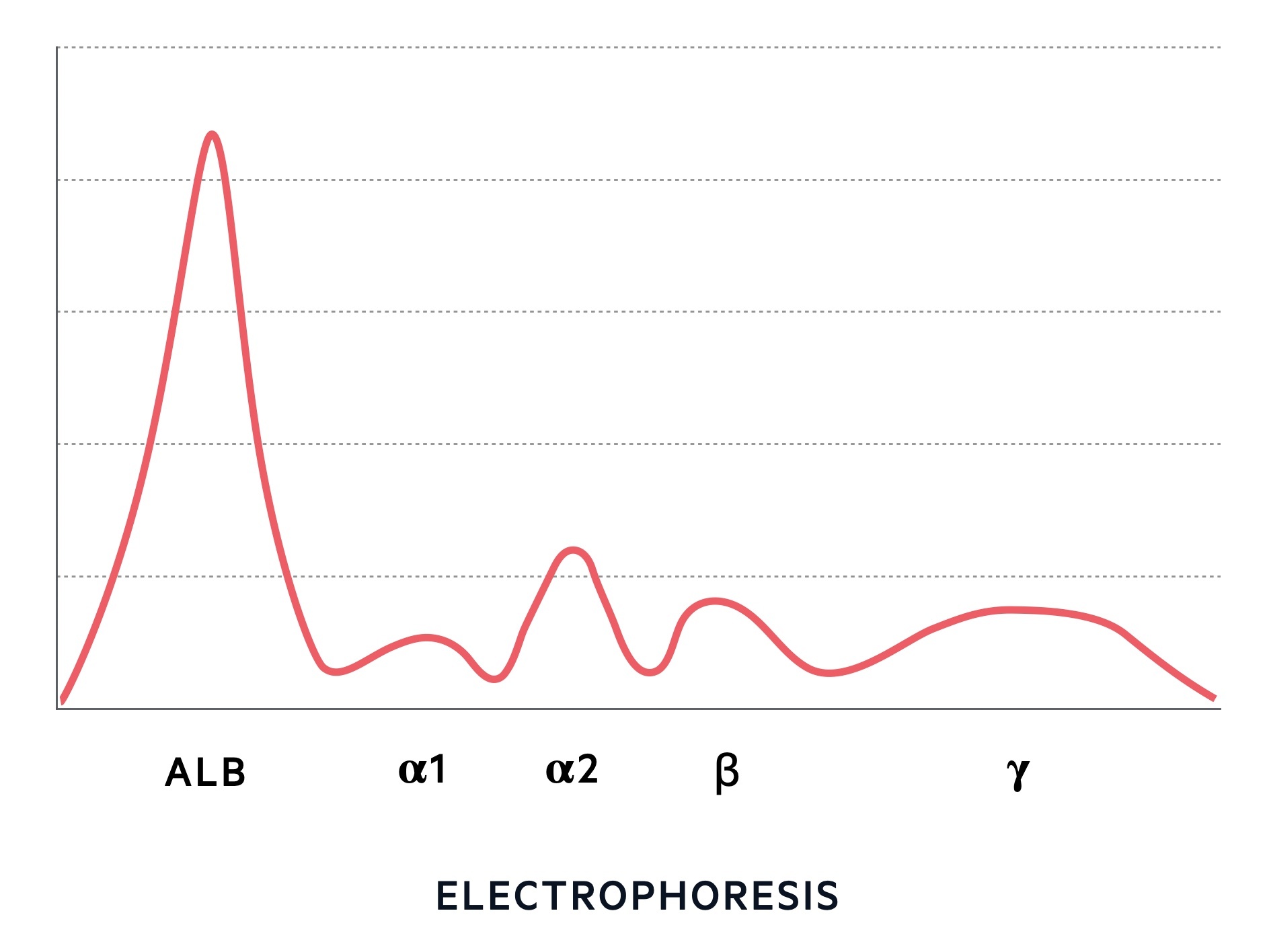
Protein electrophoresis
This is a quantitative test that separates proteins into different bands using an electric current. The distance individual proteins travel is dependent on their shape, size and electrical charge. Electrophoresis gives us characteristic band patterns including normal, polyclonal and monoclonal.
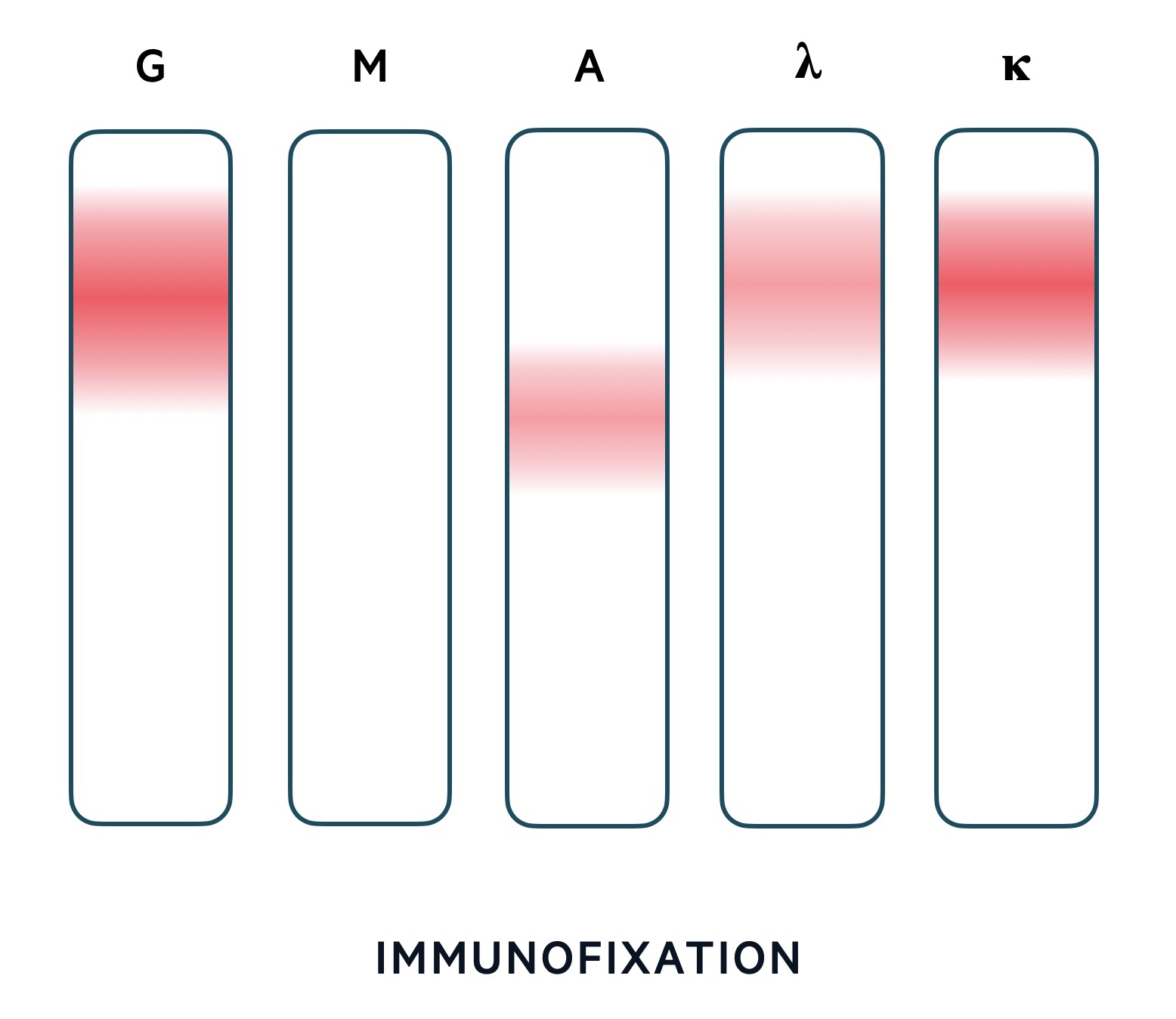
Immunofixation
Immunofixation is a qualitative test that ‘fixes’ proteins in place by using antibodies. It is important for the identification of proteins after separation by electrophoresis.
Urine electrophoresis and serum free light chains
Protein electrophoresis assumes that all myelomas secrete an intact antibody. In fact, around 20% of myelomas only secrete light chains. To help detect these myelomas we can send off serum free light chains (SFLCs) or urine for electrophoresis.
SFLCs is a newer test that looks at the amount of light chain unbound to heavy chains within the blood. Light chains are secreted in healthy individuals as plasma cells produce more light chains than heavy chains. Therefore, it is the ratio between the light chains kappa and lambda, which is the most important factor. An elevated ratio is suggestive of myeloma and needs further work-up.
Alternatively, a urine electrophoresis can be completed. Light chains within the serum may be filtered by the kidneys into the urine. Monoclonal light chains detected in the urine are known as Bence-Jones proteins. NICE recommend the use of serum protein electrophoresis and SFLCs in the work-up of suspected myeloma. Depending on the centre, urine electrophoresis may still be used in combination with the above two tests or as an alternative to SFLCs.
NOTE: A small percentage of patients with MM do not have detectable paraprotein levels (i.e. protein electrophoresis and SFLCs are negative). These patients have non-secretory myeloma.
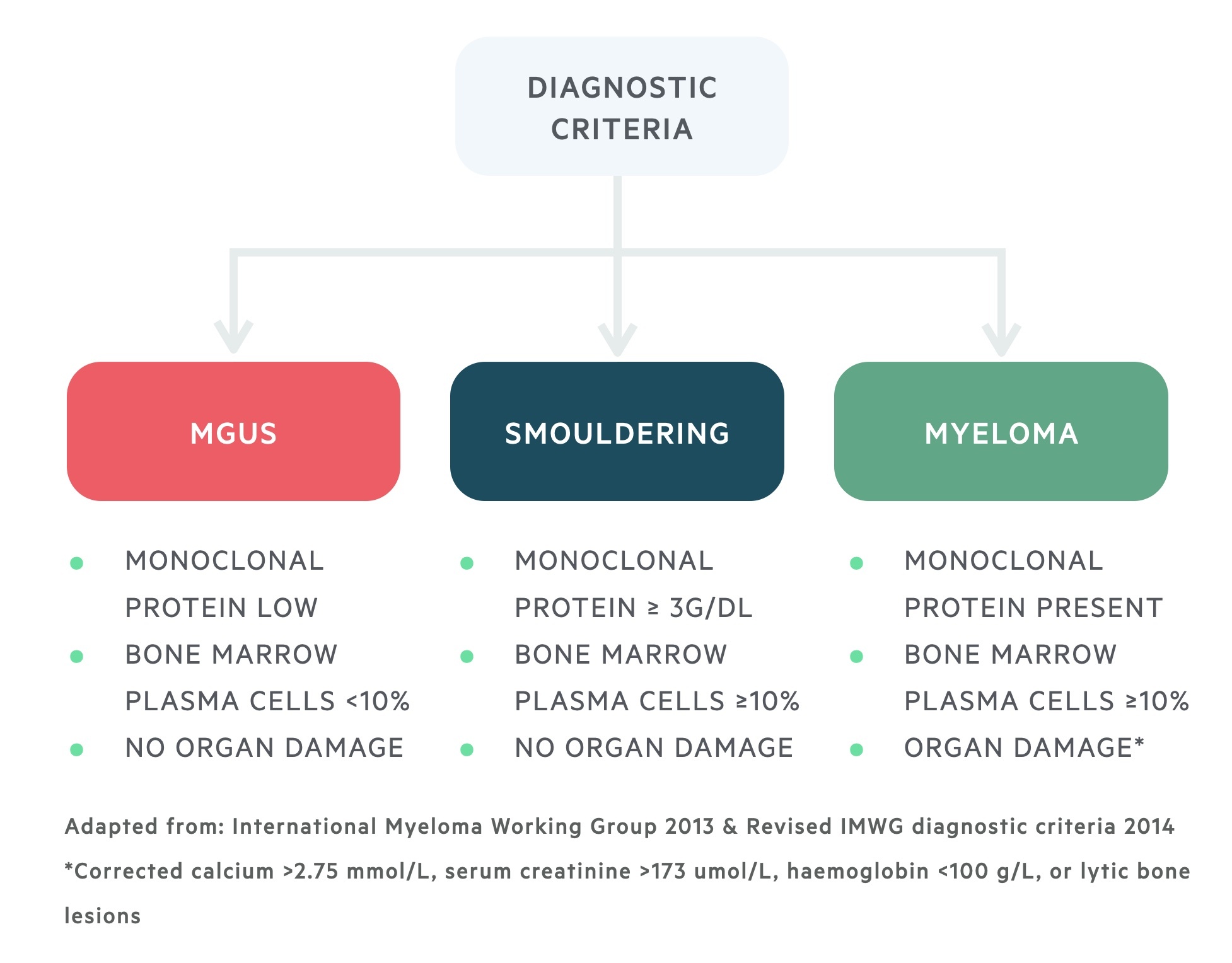
Diagnostic criteria
Diagnosis of MM involves identifying a monoclonal antibody, bone marrow analysis and assessing organ damage.
The work-up and diagnosis of MM is dependent on identification of a monoclonal antibody (sometimes referred to as M protein or paraprotein), analysis of the bone marrow to look for a population of malignant clonal plasma cells and further laboratory tests and imaging to assess for myeloma-related organ damage.
- Monoclonal antibody detection: protein electrophoresis & immunoglobulins, SFLCs +/- urine electrophoresis for Bence-Jones protein
- Bone marrow infiltration: bone marrow aspirate and trephine with cytogenetics
- Myeloma-related organ damage: FBC, U&Es, bone profile, imaging (whole body MRI or low-dose whole body CT if MRI not suitable). Skeletal survey (x-rays) only used if CT/MRI not possible
- Staging if confirmed myeloma: beta-2 microglobulin, albumin
Collectively, these investigations are important as part of the diagnostic criteria of myeloma. They help differentiate between MGUS, smouldering/asymptomatic myeloma and multiple myeloma.
Treatment principles of myeloma
MM is an incurable condition, treatments aim to increase periods of disease remission.
There are many options for the treatment of myeloma. Management depends on a patients fitness and co-morbidities, disease severity, initial response to treatment, relapse(s) and previous therapy. All patients should be discussed in an MDT specialising in myeloma and have access to psychological services, palliative care and support, specialist nurses and clinical research.
Unfortunately, there is no cure for myeloma. The aim is to induce disease remission and then maintain disease free survival for as long as possible with ongoing monitoring for disease relapse. The four key areas of management include: induction therapy, autologous stem cell transplantation (ASCT), maintenance therapy and managing relapse or refractory disease.
- Induction therapy: initial treatment option. Aim to induce remission. Usually combination of three drugs. Choice depends on high-risk features, co-morbidities and plan for ASCT. Example is VRd (Velcade - bortezomib / Revlimid - lenalidomide / dexamethasone - steroid)
- ASCT: if suitable for transplant, provides best option for long period of remission. Stem cells mobilised, harvested and stored following induction. Subsequently given high-dose chemotherapy (e.g. melphalan). Stem cells then re-infused.
- Maintenance: used to maintain disease remission as long as possible. Given post-induction or post-transplant. Choices include bortezomib or lenalidomide. Typically given until progression.
- Relapse or refractory disease: almost all patients will relapse, even if they respond to treatment. Therapy indicated if a clinical relapse or rapid rise in paraproteins. Choices include ASCT, rechallenge with previous regimen, or new therapy.
Treatment of myeloma complications
Myeloma is associated with a number of complications, which require further management.
Myeloma can lead to devastating complications due to its proliferation in bones, affect on the kidneys and neurological involvement. The management of individual complications is beyond the scope of these notes, but use the linked below for further information on these topics (albeit not specifically linked to myeloma).
- Myeloma bone disease: Bisphosphonates used for boney pain.
- Hypercalcaemia
- Cord compression
- Renal impairment
- Anaemia
Prognosis
MM is an incurable disease with a variable natural history.
Patients with myeloma will invariably relapse following treatment. Subsequent relapses are associated with reducing response to treatment. The median survival is highly variable between patients depending on response to treatment, age of onset and cytogenetic abnormalities.
Beta-2 microglobulin is often used as a prognostic tool in the International Staging System (ISS) for myeloma. This divides patients into three groups (I, II, III) based on serum beta-2 microglobulin and albumin levels, which predicts the median survival.
- Stage I: median survival 62 months
- Stage II: median survival 44 months
- Stage III: median survival of 29 months
Other factors associated with a worse prognosis include high plasma cell counts, high levels of monoclonal antibody in blood/urine or development of complications (e.g. diffuse multiple bone lesions, marked anaemia, hypercalcaemia and renal impairment).
Last updated: November 2021
Have comments about these notes? Leave us feedback