Haemolytic anaemia
Notes
Overview
Haemolysis refers to the destruction of red blood cells (RBCs), which is broadly defined as a reduction in the life span below 100 days (normal 110-120 days).
Haemolytic anaemia is defined as anaemia secondary to reduced survival of RBCs. They have a varied aetiology, as is the clinical presentation; together they represent approximately 5% of all anaemias.
Regardless of the underlying cause, if erythropoiesis within the bone marrow cannot keep pace with the destruction of RBCs anaemia will ensue. Mild haemolysis may be completely asymptomatic whereas severe, acute haemolysis will lead to cardiopulmonary decompensation.
Classification
Haemolytic anaemia can be classified as inherited or aquired.
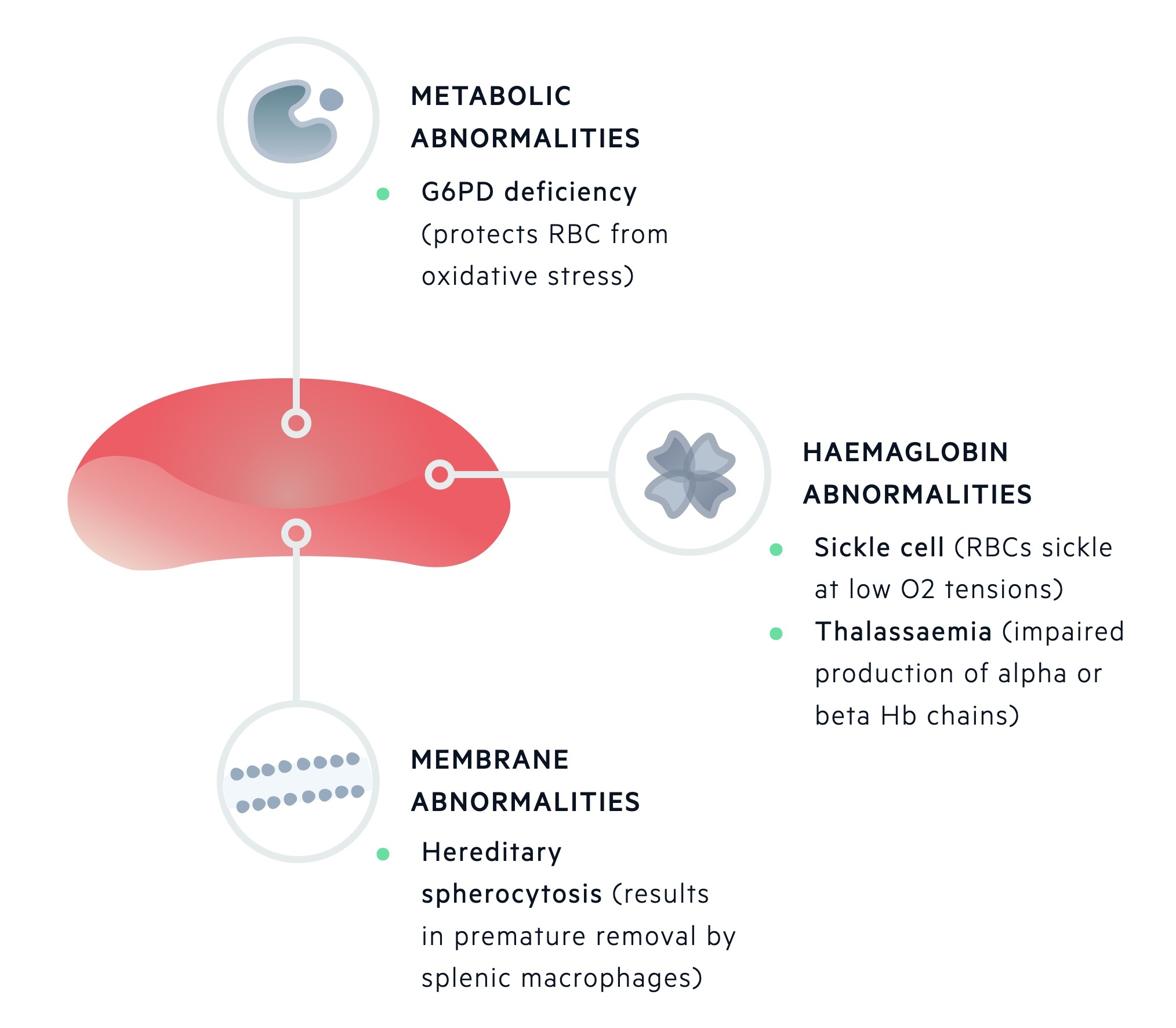
Inherited haemolytic anaemias
Inherited defects may affect one or more of the components of a red blood cell. During maturation, RBCs lose their nucleus, RNA and mitochondria.
This leaves mature RBCs with three main components:
- Red cell membrane
- Metabolic machinery
- Haemoglobin
Inherited haemolytic anaemias can be divided based upon these primary sites of pathology.
Acquired haemolytic anaemias
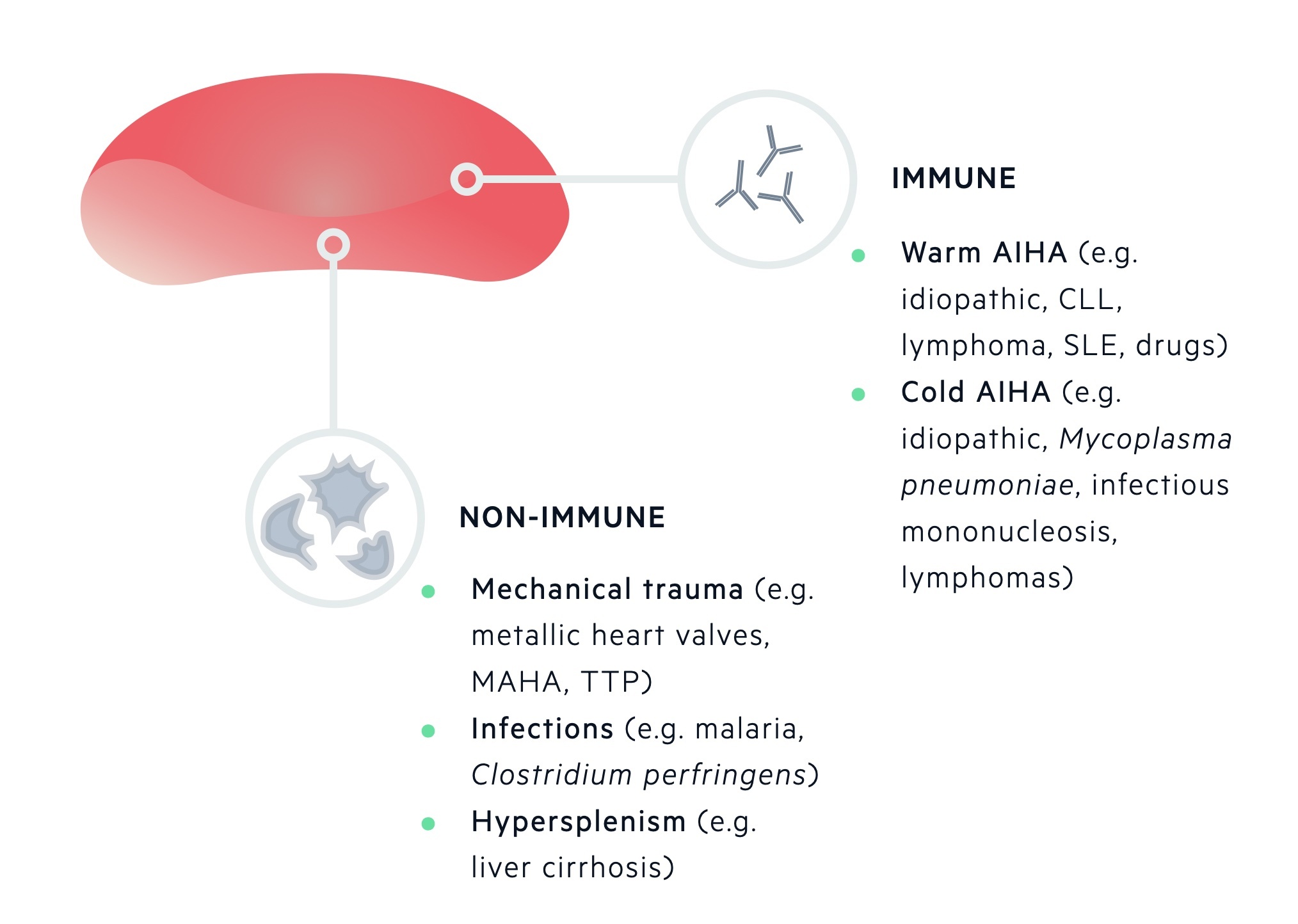
Acquired causes of RBC destruction are categorised by mechanism of destruction (immune or non-immune mediated).
Immune-mediated destruction of RBCs occurs due to antibodies targeted against components of the RBC membrane. This may lead to fixing of complement and subsequent phagocytosis by macrophages. One of the more well-known causes is autoimmune haemolytic anaemia (AIHA). It is characterised by either warm-reactive antibodies or cold haemagglutinins.
Non-immune mediated destruction of RBCs can occur by a number of mechanisms which include mechanical distortion from abnormal surfaces (e.g. prosthetic heart valve), passing through abnormal intravascular fibrin strands (e.g. microangiopathic haemolytic anaemia), infections, drugs or an overactive spleen (e.g. hypersplenism).
Clinical features
Clinical features can be grouped into those of anaemia, haemolysis & the underlying aetiology.
Symptoms
- Fatigue
- Weakness
- Paraesthesia
- Dyspnoea
- Gastrointestinal symptoms (e.g. nausea, dyspepsia)
- Weight loss
Signs
- Atrophic glossitis
- Pallor
- Fever
- Splenomegaly
- Evidence of underlying disease
Haemolysis
- Jaundice
- Abdominal pain (e.g. gallstones)
- Dark urine (e.g. haemoglobinuria secondary to intravascular haemolysis)
Underlying aetiology
- Neurological signs (e.g. TTP)
- Splenomegaly
Investigations
Diagnostic work-up for patients with suspected haemolytic anaemia should begin with a haemolytic screen.
The haemolytic screen includes a standard set of blood tests that help to confirm the presence of haemolysis and may suggest an underlying diagnosis. A standard haemolytic screen consists of:
- FBC (inc. reticulocyte count)
- Blood film
- LDH
- LFTs (bilirubin)
- Serum haptoglobin
- Additional tests
FBC
The full blood count is essential to diagnose anaemia and provide evidence of other parameters such as white blood cell and platelet counts.
Using the mean corpuscular volume (MCV), we may delineate the cause of anaemia based on the morphology of the red blood cells (macro-, normo- and microcytic). Haemolytic anaemia is traditionally causes as a normocytic anaemia.
The reticulocyte count is important in the diagnosis of haemolytic anaemia. Reticulocytes are immature red blood cells that are slightly larger and contain remnant nuclear material. The normal percentage of reticulocytes in the peripheral blood is 1-2%. In the absence of bone marrow disease, an increase in RBC destruction leads to the release of EPO, which stimulates the bone marrow to produce more erythrocytes, subsequently increasing the percentage or reticulocytes.
Blood film
The peripheral blood film is important to assess the morphological characteristics of erythrocytes and can detect concomitant abnormalities such as haematological malignancies.
Typical erythrocyte morphologies associated with haemolysis may include:
- Spherocytes (e.g. hereditary spherocytosis)
- Schistocytes (e.g. microangiopathic haemolytic anaemia)
- Sickle cells (e.g. sickle cell disease)
Additional tests
Lactate dehydrogenase (LDH) is a non-specific marker of cell damage and turnover. LDH is sensitive for the presence of haemolysis, but it is a not specific.
Liver function tests (LFTs) are essential to assess the level of bilirubin. In haemolysis there is an increased production of bilirubin (a breakdown product of haemoglobin). If the liver is unable to completely compensate (by conjugating and excreting the bilirubin) a mild unconjugated hyperbilirubinaemia may be seen.
Haptoglobin is a plasma protein, which binds to free haemoglobin within the blood. In the presence of intravascular haemolysis, free haemoglobin is mopped up by haptoglobin. The haemoglobin-haptoglobin complex is removed by the liver leading to a decrease in the level of haptoglobin. Importantly, haptoglobin is an acute phase reactant and as such levels my be raised despite significant haemolysis.
Following a haemolytic screen, further investigations may be completed to try and detect the underlying cause. These may include a direct anti-globulin test (DAT), haemoglobin electrophoresis and sickle cell screening. Other tests suggestive of abnormal intravascular haemolysis include free urinary haemoglobin and urinary haemosiderin tests.
Inherited haemolytic anaemias
Inherited haemolytic anaemias can be grouped depending on the primary site of pathology being the erythrocyte membrane, metabolic machinery or haemoglobin molecule.
Erythrocyte membrane
The erythrocyte membrane is essential to allow RBCs to undergo deformation as they pass through the capillary bed and then recoil back into shape. It is also needed to regulate the entry of water and important cations (e.g. Na+, K+, Ca2+, and Mg2+).
Unstable / missing erythrocyte membrane proteins may be seen in hereditary spherocytosis or elliptocyosis. Hereditary spherocytosis is the most common inherited form of haemolysis, which is usually transmitted in an autosomal dominant pattern. The condition is characterised by mutations that lead to defects within the RBC membrane resulting in cytoskeleton instability.
It is associated with a low haemoglobin concentration, raised reticulocyte count and spherocytes seen on blood film, which are smaller and denser erythrocytes that have a sphere-shape as opposed to the normal bi-concave erythrocytes.
Patients with hereditary spherocytosis are usually unaffected, but if treatment is required, it typically involves a splenectomy that dramatically increases the red blood cell count.
Metabolic machinery
The metabolic apparatus is essential to generate energy in the form of ATP for the transfer of cations, function of 2,3-BPG and protection against oxidative stress.
Defects in the metabolic apparatus can predispose RBCs to oxidative damage. One of the most common causes worldwide is glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency). This is an X-linked recessive disorder, which may lead to episodes of haemolysis in the presence of oxidative stressors.
Normally, G6PD is important for the generation of NADPH, which is a reducing agent that helps to scavenge oxidative metabolites that may otherwise damage the RBC.
The disease has a high prevalence in patients of African, Asian and Mediterranean descent, but usually, no treatment is required apart from the avoidance of oxidative stressors (e.g. fava beans, types of henna and numerous drugs).
Haemoglobin molecule
Haemoglobin is the major protein of erythrocytes, which is essential for the transfer of oxygen. Normal adult haemoglobin (HbA) is composed of two beta and two alpha global chains.
Inherited defects in these important genetic regions can lead to alteration in the composition of haemoglobin, which can have an effect on membrane stability and erythrocyte survival. Important conditions include:
- Sickle cell disease is an autosomal recessive disease that occurs due to a point mutation within the beta-globin gene. The resultant haemoglobin (HbS) is 50x less soluble than HbA and is at risk of polymerising under low-oxygen tension, which further damages the RBC.
- Alpha-thalassaemia and beta-thalassaemia are characterised by a genetic deficiency of alpha and beta-globin chains, respectively. Both conditions are characterised by the absent or reduced production of normal globin chains leading to an imbalance in chain production, abnormal erythropoiesis and defective erythrocytes.
Acquired haemolytic anaemias
Acquired haemolytic anaemias can be due to immune-mediated or non-immune mediated destruction.
Immune-mediated
Occurs due to the binding of antibodies to components of the erythrocyte membrane, which can lead to fixing of complement and phagocytosis by macrophages.
Antibodies that bind to the erythrocyte membrane may be alloantibodies or autoantibodies.
- Alloantibodies are antibodies produced by one individual that will react with antigens of another individual of the same species. May be seen in haemolytic transfusion reactions and haemolytic disease of the newborn.
- Autoantibodies, which are generated against components of the individuals own tissue, may be seen in AIHA. AIHA is further divided depending on the type of autoantibody present being ‘warm’ or ‘cold’.
Warm AIHA is associated with an antibody reaction against erythrocytes at higher temperatures (e.g. > 37°), which then leads to agglutination. Warm AIHA may be idiopathic, or associated with immune dysfunction secondary to infection, chronic inflammation or malignancy. Warm AIHA may be associated with infections such as HIV and EBV, inflammatory disorders such as SLE, or malignancies such as chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma.
Cold AIHA is associated with a reaction against erythrocytes at lower temperatures (e.g. < 32°), which then leads to agglutination. Cold AIHA may be idiopathic or associated with systemic diseases such as lymphoma, Mycoplasma pneumoniae infection and infectious mononucleosis.
Non-immune mediated
Non-immune mediated destruction of erythrocytes can occur due to a number of different mechanisms.
- Mechanical trauma - due to heart and large blood vessel pathology (e.g. prostheses).
- Microangiopathic haemolytic anaemia (e.g. HUS, TTP, DIC).
- Burns
- Infections
- Drugs & chemicals
- Hypersplenism
RBCs may become mechanically damaged from the impact on abnormal surfaces such as metallic heart valves or as they pass through abnormal, intravascular fibrin strands that may be seen in microangiopathic haemolytic anaemias.
Finally, hypersplenism is a term that refers to the reduction in RBCs, platelets and granulocytes in association with splenomegaly, regardless of cause.
Last updated: March 2021
Have comments about these notes? Leave us feedback